مرض نادر إلى حد ما ، يحدث في 1 من 2 مليون ، متلازمة الرجل الحجري ، المعروف طبيا باسم خلل التنسج الليفي العظمي (FOP) ، هو مرض وراثي ناتج عن طفرة في الجينات تسمح للنسيج الضام المصاب بالتحول إلى عظم. في الأشخاص الذين يعانون من مرض الحصيات ، يتم إنشاء هيكل عظمي جديد. يبدأ التنكس بعظام الرقبة وينتشر إلى أسفل مؤثرًا على جميع الهياكل ، بما في ذلك العجز.
في السابق ، تم الإبلاغ عن عدة حالات لهذا المرض. يمكن العثور على الهياكل العظمية التي تظهر تعظم النسيج الضام في متحف موتر للتاريخ الطبي في فيلادلفيا بالولايات المتحدة الأمريكية ولندن. في الواقع ، هم جزء من مجموعة متحف Hunterian في الكلية الملكية للجراحين.
تبدأ متلازمة الرجل الحجري في الظهور في سن مبكرة ، على مر السنين تتطور المزيد والمزيد من العظام. يتمكن معظم المرضى من العيش لمدة 40 عامًا تقريبًا ، حيث تزداد احتمالية الوفاة نتيجة مشاكل التنفس بعد هذا العمر. تشكل الإصابات الرياضية أو إصابات السقوط أو حتى الإجراءات الطبية الغازية خطورة على هؤلاء الأشخاص ، لأنها يمكن أن تسبب التهابًا شديدًا في العضلات والأوتار والأربطة ، وبدلاً من ذلك ، على الرغم من الشفاء ، تتشكل العظام لتحل محل الأنسجة الضامة المصابة.
الأسباب
متلازمة الرجل الحجري هي اضطراب وراثي جسمي سائد ناتج عن طفرة في جين ACVR 1 (نوع مستقبلات أكتيفين 1). في معظم الحالات ، يكون هذا الاضطراب عفويًا ، وتحدث طفرات جديدة في حالة عدم وجود تاريخ عائلي للمرض. يمكن أن تظهر نتيجة التعرض للإشعاع المؤين ، مواد كيميائيةوالمخدرات والالتهابات. يمكن أن يحدث هذا التعرض في الرحم أو بعد الولادة بقليل. ومع ذلك ، تم تسجيل العديد من حلقات انتقال الجينات من الأبوين إلى الأبناء في التاريخ الطبي. في ظل هذه الظروف ، يكون أليل واحد (نسخة) متغيرة من الجين الأبوي كافياً لظهور المرض.
في ظل الظروف العادية ، يعمل جين ACVR 1 كمُحَوِّل يتحكم في نمو وتكاثر خلايا العضلات والأربطة والأنسجة الضامة الأخرى. يقوم بتشفير البروتين التكويني للعظام VMP 1 ، الذي يتحكم في التعظم الطبيعي ونضج عظام الهيكل العظمي. عندما تحدث طفرة بسبب إصابة أو عدوى فيروسية في العضلات والأنسجة الضامة ، يتم تنشيط الجين باستمرار ، يليه إطلاق البروتينات المعيبة. هذا يؤدي إلى ترسب الخلية أنسجة العظامفي المناطق المتضررة ، النمو المفرط للعظام وانصهار المفاصل والعظام.
أعراض
أعراض متلازمة الرجل الحجري مميزة تمامًا وتتطور طوال الحياة. وتشمل هذه:
- يعتبر وجود أصابع كبيرة مشوهة أول علامة على هذا الاضطراب. يعد هذا أيضًا عرضًا مهمًا لأنه يساعد الأطباء على التمييز بينه وبين الحالات العضلية الهيكلية الأخرى المماثلة.
- صعوبة في الحركة وتيبس المفاصل نتيجة اندماجها مع العظام حديثة التكوين.
- - صعوبات في الأكل نتيجة التحام عظام الفك. بسبب سوء التغذية ، كقاعدة عامة ، تحدث الأمراض المصاحبة. يؤدي اندماج مفصل الفك أيضًا إلى مشاكل في الكلام.
- مع تكوين عظام جديدة في المنطقة صدر، يصبح توسعها أكثر تعقيدًا ، مما يؤدي إلى مشاكل في التنفس. يموت معظم مرضى متلازمة الرجل الحجري بسبب فشل الجهاز التنفسي والالتهاب الرئوي.
- الأشخاص الذين يعانون من هذا المرض لديهم حدبة مميزة في الظهر وانحراف العمود الفقري إلى الجانب وصعوبة في الحركة وعدم القدرة على أداء مهام معينة. يبدأ الجسم في الظهور وكأنه تمثال صلب. في أغلب الأحيان ، في العقد الثاني أو الثالث من العمر ، يصبح المرضى طريح الفراش تمامًا.
كيفية التعامل معها
حتى الآن ، لا يوجد علاج لمتلازمة الرجل الحجري. ومع ذلك ، فإن استخدام جرعات عالية من الكورتيكوستيرويدات يمكن أن يساعد في تقليل الالتهاب في العضلات والأنسجة الضامة ، وبالتالي تأخير تكوين العظام الجديدة. ينصح المرضى عمومًا بتجنب السقوط والإصابة والرياضات التي تتطلب الاحتكاك الجسدي.
اختبار الفترة المحيطة بالولادة للأطفال المولودين بهذا العيب ليس إجراءً روتينيًا. ومع ذلك ، يمكن تأكيد التشخيص نفسه عن طريق الاختبارات الجينية.
متلازمة الرجل الحجري في فتاة مراهقة

لسنوات ، كانت سيني نيموك ، فتاة جميلة تبلغ من العمر 18 عامًا من شمال كنسينغتون في لندن ، تكافح مرضًا وراثيًا نادرًا. تم تشخيص سيني لأول مرة بمتلازمة الرجل الحجري في سن 12. نتيجة لسقوط صغير ، تم استبدال المنطقة الملتهبة على الظهر تدريجياً بأورام العظام وتسبب في ألم شديد. اندمج العنق والعمود الفقري في واحد ، مما منع الفتاة من رفع ذراعيها فوق خصرها. تخيم على حياتها اليوم مخاوف مستمرة من السقوط والإصابات ، حيث سيؤدي ذلك بالتأكيد إلى إنبات عظام جديدة وتفاقم الحالة الحالية.
على الرغم من هذا التشخيص الصعب ، لا يزال سيني يقضي الوقت مع أصدقائه. إنها تحب الطبخ والتسوق ، ومثل أي فتاة مراهقة عادية ، تحب المكياج. لا تترك عائلتها الأمل في أن العلماء سيظلون قادرين على فهم هذه العملية المرضية المعقدة وخلق علاج لمتلازمة الرجل الحجري.
سوروكينا يوليا سيرجيفنا
كيف يظهر التهاب الجلد التأتبي في مرحلة الطفولة؟ تظهر العلامات الأولى للمرض عند معظم الأطفال في سن الرضاعة. يرتبط مظهرها ، كقاعدة عامة ، بإدخال الخلطات الاصطناعية وحليب البقر والبيض والأسماك وبعض الحبوب في النظام الغذائي. يظهر احمرار ، بثور على الوجه والجذع والذراعين والساقين للأطفال ، ويبتل الجلد ، أو على العكس من ذلك ، يجف ويتقشر. يصبح الأطفال مضطربين ، ولا ينامون جيدًا. إذا لم تفوت اللحظة ولجأت إلى مساعدة طبيب الأمراض الجلدية ، فيمكن إيقاف التهاب الجلد التحسسي. العمر حتى 3 سنوات هو أكثر الأوقات امتنانًا للعلاج. في هذه الفترة من الطفولة ، من الممكن تحقيق أقصى درجات الاحتمال
تظهر العلامات الأولى للمرض عند معظم الأطفال في سن الرضاعة. يرتبط مظهرها ، كقاعدة عامة ، بإدخال الخلطات الاصطناعية وحليب البقر والبيض والأسماك وبعض الحبوب في النظام الغذائي. يظهر احمرار ، بثور على الوجه والجذع والذراعين والساقين للأطفال ، ويبتل الجلد ، أو على العكس من ذلك ، يجف ويتقشر. يصبح الأطفال مضطربين ، ولا ينامون جيدًا. إذا لم تفوت اللحظة ولجأت إلى مساعدة طبيب الأمراض الجلدية ، فيمكن إيقاف التهاب الجلد التحسسي. العمر حتى 3 سنوات هو أكثر الأوقات امتنانًا للعلاج. في هذه الفترة من الطفولة ، من الممكن تحقيق أقصى درجات الاحتمال
انقطاع "مسيرة" التأتب.
في سن 6 - 7 و 12 - 14 سنة ، من الممكن حدوث تفاقم في عملية الجلد. يتم استفزازه عن طريق غبار المنزل وشعر الحيوانات الأليفة وحبوب اللقاح والبكتيريا والعفن والمواد المسببة للحساسية الغذائية في الخلفية. تلعب المواقف العصيبة دورًا مهمًا ، وانتهاك الروتين اليومي ، وإثقال كاهل الطفل بالدراسات ، والدروس في العديد من الدوائر. ينتقل الالتهاب إلى ثنايا الذراعين والساقين والرقبة. يصبح الجلد أكثر جفافاً وسميكاً وتتشكل قشور في موقع الخدش.
في بعض الناس ، تكون مظاهر التأتب في الطفولة غير مهمة ولا يلاحظها الآباء وأطباء الأطفال ، ولكنها تظهر مرة أخرى في مرحلة البلوغ ، على ما يبدو "لأول مرة". يحدث التهاب الجلد في كثير من الأحيان في ثنايا الأطراف والأصابع ومفاصل الكاحل والوجه والرقبة. قد يكون الطفح الجلدي باكيًا أو يشبه الإكزيما أو جافًا. يجمع بين الحكة القوية والتأثيرات المتبقية في شكل جلد خشن سميك وتقشير وجفاف.
مسيرة تأتبيةكقاعدة عامة ، فإنها تتطور في وقت متأخر عن مظاهر التهاب الجلد التأتبي. في السنوات الاخيرةنتحدث عن ما يسمى "مسيرة التأتبي". ما هذا؟ تعني المسيرة التأتبية أن "أهبة" الأطفال يمكن أن تكون بمثابة المرحلة الأولية لتطور أشكال أخرى أكثر حدة من الحساسية - الربو القصبي وحمى القش (حساسية من حبوب اللقاح النباتية) والحساسية تجاه الطعام والدواء وحساسية الأنف (سيلان الأنف) ) والتهاب الملتحمة (التهاب العين) وآلام المفاصل والصداع النصفي. علاوة على ذلك ، يمكن أن تتغير هذه الأمراض في فترات مختلفة من حياة شخص واحد. على سبيل المثال ، كان هناك طفح جلدي على الجلد ، ثم ذهب كل شيء ، وظهر داء اللقاح أو الصداع النصفي. من خلال التحكم في مسار التهاب الجلد التأتبي ، يأمل الأطباء والعلماء في منع "المسيرة التأتبية".
التأتب في الميراث؟التهاب الجلد التأتبي والتأتب هما حالتان خلقية وراثية. هذا يعني أن هناك عوامل وراثية تحدد التأتب. تتم الآن دراسة هذه الآليات بعناية لتكون بمثابة أساس لتطوير علاجات جديدة للتأتب في المستقبل.
لم يتم تحديد جميع طرق وراثة التهاب الجلد التأتبي بشكل نهائي. نعلم أنه إذا كان أحد الوالدين أو الأقارب المقربين للطفل مصابًا بالتهاب الجلد التأتبي أو الربو أو التهاب الأنف التحسسي ، فإن الطفل نفسه أو نفسها لديه فرصة بنسبة 50٪ للإصابة بالتهاب الجلد التأتبي. في الوقت نفسه ، لا يعاني أقارب 30 ٪ من المصابين بالتهاب الجلد التأتبي من أي مظاهر للحساسية.
الوراثة التأتبية ليست حكمًا يقضي على الشخص مدى الحياة أو يعاني من أعراض الحساسية لبعض الوقت. من الممكن أن تكون "تأتبية" وليس لديك طفح جلدي أو حكة.
ظهور شخص مصاب بالتأتبالجلد المصاب بالتهاب الجلد التأتبي جاف. يُغطى جلد الجذع والأسطح الباسطة للذراعين و / أو الساقين "ببثور" صغيرة لامعة بلون اللحم. على الأسطح الجانبية للكتفين ، والمرفقين ، وأحيانًا في منطقة مفاصل الكتف قد يكون هناك حطاطات صغيرة كثيفة قرنية ("المبشرة"). في سن أكبر ، يتلألأ الجلد بوجود بقع داكنة وبيضاء. في كثير من الأحيان يتم تحديد بقع بيضاء في منطقة الخدين.
خلال فترة الهدوء ، قد يكون الحد الأدنى من مظاهر التهاب الجلد التأتبي قشاريًا قليلاً أو جلد سميك قليلاً أو حتى تشققات في منطقة التعلق بالأذن. بالإضافة إلى ذلك ، يمكن أن تكون هذه العلامات هي التهاب الشفة (تشققات وجفاف الشفاه) ونوبات متكررة (تشققات عميقة في زوايا الفم) وتشق متوسط في الشفة السفلية واحمرار الجلد الجاف في الجفون العلوية. يمكن أن تكون الهالات السوداء حول العين وشحوب البشرة الترابية مؤشرات مهمة على الاستعداد التأتبي.
كيف يؤثر التهاب الجلد التأتبي على علم النفس؟لا يقتصر التأثير النفسي-العاطفي على التجارب العشوائية وقصيرة المدى بسبب تفاقم الطفح الجلدي. المظاهر غير المنضبطة لالتهاب الجلد التأتبي تؤدي باستمرار إلى تعقيد حياة "التأتبي". تسبب الحكة عدم الراحة ، وتؤدي أحيانًا إلى الأرق والتهيج والاكتئاب وزيادة التعب. يجب إخفاء الطفح الجلدي في المناطق المفتوحة من الجسم عن الآخرين ، وعليك أن تتذكر طوال الوقت أنه لا يمكنك فعل شيء ما ، أو أنه سيتسبب مرة أخرى في الحكة والطفح الجلدي.
التهاب الجلد التأتبي له تأثير قوي على طريقة حياة الشخص بالكامل ونظرته للعالم ، وأحيانًا يجعله "مختلفًا" حقًا ، حيث ينظر إلى العالم بطريقة مختلفة ، حيث يوجد العديد من الأعداء الذين يحتمل أن يكونوا خطرين ومسببين للحساسية لدرجة أن البقية عادة يسامح. يُعتقد أن "التأتبيين" أكثر عرضة للملاحقات الفكرية والتحليل المدروس والدقيق لما يحدث حولهم ، وأكثر حساسية وانسحابًا.
لا تؤثر مظاهر التهاب الجلد التأتبي على نفسية المريض فحسب ، بل تؤثر أيضًا على الأسرة بأكملها والأشخاص المحيطين به وعلاقاتهم المتبادلة.
ماذا تفعل التأتبي؟ يوجد الكثير منا "التأتبيون": 1-3٪ من جميع الأوروبيين البالغين و 10-20٪ من أطفال المدارس. وإليك كيف اتضح: "إنها مسألة حياة!" يمكننا التغلب على الإجهاد والتهاب الجلد التأتبي.
يوجد الكثير منا "التأتبيون": 1-3٪ من جميع الأوروبيين البالغين و 10-20٪ من أطفال المدارس. وإليك كيف اتضح: "إنها مسألة حياة!" يمكننا التغلب على الإجهاد والتهاب الجلد التأتبي.
للتغلب على التوتر العاطفي والمشاكل الاجتماعية ، أنت بحاجة إلى الثقة بالنفس والقدرة على التحكم في حالة بشرتك. أنت بحاجة إلى مناقشة مفتوحة وسرية لحالتك مع الأصدقاء والأقارب والأطباء وغيرهم من الأشخاص الذين يعانون من نفس المشكلة.
للحفاظ على حالة الهدوء ، يلزم عناية خاصة بالبشرة ؛ طور أطباء الجلد منتجات خاصة - المطريات. العديد من العلامات التجارية المعروفة لمستحضرات التجميل لديها مجموعة من المنتجات الخاصة بالتأتب
الجلد: أفين ، لاروش بوزيه ، إلخ. مع الاستخدام المستمر للهلام وكريمات بشرة الجسم من هذه السلسلة ، نقوم باستعادة الحاجز الواقي للجلد والتخلص من ردود الفعل غير الكافية.
ما الذي يمكن استخدامه أيضًا بشكل مستقل للتأتب؟ بالطبع ، المواد الماصة! هذه هي المواد التي تؤخذ عن طريق الفم ، وتربط السموم ، وصلات مختلفةوإخراجها من الجسم بشكل طبيعي. كواحد من المواد الماصة التي أعرفها لفترة طويلة. يشار إلى المواد الماصة في علاج التهاب الجلد التأتبي. من الصعب على المرضى المصابين بالحساسية الشديدة شرب كوب من الماء مع أقراص ماصة في جرعة واحدة. المسحوق الناعم هو الأنسب للأطفال ، حيث يسهل تخفيفه بكمية قليلة من الماء ، على عكس مثيلاته ذات الحبيبات الخشنة ، مما يسهل تناوله. ثم يمكنك شرب كوب من الماء خلال نصف ساعة. بعد دورة مدتها أسبوعين ، غالبًا ما تختفي علامات التفاقم التي بدأت ، حتى بدون استخدام علاج محدد. تؤخذ المادة الماصة التأبتية عند البالغين أثناء المرحلة الحادة من التهاب الجلد وللوقاية قبل التفاقم الموسمي.
أين يجب أن يعالج مريض التهاب الجلد التأتبي؟من الضروري أن تتم ملاحظته وعلاجه من قبل طبيب الأمراض الجلدية نفسه. إذا لزم الأمر ، يقوم طبيب الأمراض الجلدية بتعيين الاستشارات مع الأطباء المتخصصين: طبيب أعصاب ، وأخصائي أمراض الجهاز الهضمي ، وأخصائي الحساسية ، وأخصائي أمراض العمود الفقري ، وما إلى ذلك. هذه هي الطريقة الوحيدة لضمان استمرارية أنواع العلاج المختلفة ، لتتبع رد فعل المريض تجاههم.
لسوء الحظ ، يتعين علينا التعامل مع المرضى الذين ، بعد أن لم يكملوا دورة العلاج الموصوفة ، يبحثون عن علاجات خارجية جديدة ، وانتقل من طبيب إلى آخر ، وفي كل مرة ، لم يحصلوا على التأثير المتوقع ، يعانون من ضغوط إضافية ، يتبعها عن طريق تفاقم المرض. لا يجب أن تداوي نفسك. هذا مرض خطير يمكن أن يؤدي إلى مضاعفات خطيرة ، وكما قيل ، إلى تطور أشكال أخرى من "التأتب". للسيطرة على المرض ومنع المضاعفات ، يجب اتباع توصيات طبيب الأمراض الجلدية.
أندر الأمراض في العالم
كل من رجال الكهوف البدائيين والعلماء اللامعين المعاصرين - لطالما كافحت البشرية وتكافح مع العديد من الأمراض. هناك مجموعة من الأمراض الوراثية التي يصعب تجنبها إذا كان والداك مصابين بها ؛ بعض الأمراض هي نتاج طفرات جينية غير متوقعة. مجموعة كبيرة من الأمراض هي نتيجة تكاثر وازدهار الكائنات المجهرية التي استقرت داخل جسم الإنسان.
متلازمة جيلبرت
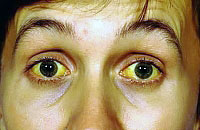
![]() متلازمة جيلبرت (GS) هي مرض وراثي يؤدي إلى ضعف استقلاب البيليروبين ، والذي يمكن أن يثير فرط بيليروبين الدم الحميد غير المقترن. يتأثر استقلاب البيليروبين بسبب نقص الجلوكورونيل ترانسفيراز ، وهو إنزيم خاص بالكبد. يؤدي هذا إلى زيادة مستوى البيليروبين غير المرتبط في الدم وحدوث اليرقان. في متلازمة جيلبرت ، كقاعدة عامة ، لا تشكل تهديدًا على الجنين. يزيد وجود SF6 من خطر الإصابة بأمراض حصوة المرارة.
متلازمة جيلبرت (GS) هي مرض وراثي يؤدي إلى ضعف استقلاب البيليروبين ، والذي يمكن أن يثير فرط بيليروبين الدم الحميد غير المقترن. يتأثر استقلاب البيليروبين بسبب نقص الجلوكورونيل ترانسفيراز ، وهو إنزيم خاص بالكبد. يؤدي هذا إلى زيادة مستوى البيليروبين غير المرتبط في الدم وحدوث اليرقان. في متلازمة جيلبرت ، كقاعدة عامة ، لا تشكل تهديدًا على الجنين. يزيد وجود SF6 من خطر الإصابة بأمراض حصوة المرارة.
تظهر أعراض متلازمة جيلبرت بشكل أكثر وضوحًا تحت تأثير الإجهاد والمجهود البدني وسوء التغذية وبعد الأمراض الفيروسية وتعاطي الكحول وبعض الأدوية. يتميز مرض جيلبرت بما يلي:
- فقد القوة؛
- اصفرار الصلبة والأغشية المخاطية بدرجات متفاوتة (لا يلاحظ دائما اصفرار الجلد) ؛
- ألم في الكبد.
- - ارتفاع مستويات البيليروبين في الدم.
- اضطراب في المعدة وهضم مؤلم.
متلازمة جيلبرت خلقية ، وفي هذه الحالة تظهر الأعراض بين سن 12 و 30 سنة. النوع الثاني من المتلازمة هو فرط بيليروبين الدم بعد التهاب الكبد ، والذي يتجلى بعد التهاب الكبد الفيروسي. في الحالة الثانية ، من الضروري إجراء التشخيص التفريقي حتى لا يتم الخلط بين GS والتهاب الكبد المزمن.
تشخيص متلازمة جيلبرت
لإجراء الدراسات التشخيصية والتخطيط للتدابير العلاجية ، من الضروري الاتصال بطبيب عام وطبيب وراثة وأخصائي أمراض الدم وأخصائي أمراض الجهاز الهضمي (أخصائي أمراض الكبد). في حالة الاشتباه في متلازمة جيلبرت ، بالإضافة إلى أخذ سوابق المريض والفحص البدني ، يتم وصف طرق التشخيص التالية:
- - مع SF ، لوحظ زيادة في الهيموغلوبين (> 160 جم / لتر) ، ومن الممكن حدوث تكوُّن شبكي وانخفاض في الاستقرار التناضحي لكريات الدم الحمراء.
- التحليل البيوكيميائي للدم - يمكن أن يصل البيليروبين إلى 6 مجم / ديسيلتر ، لكنه لا يتجاوز عمومًا حد 3 مجم / ديسيلتر. تظل المعلمات التي تحدد وظائف الكبد طبيعية. قد يزيد الفوسفاتيز القلوي.
- PCR هو علامة وراثية لـ SF ، يتكرر عدد TA في سلسلة المروج لجين UGT1A1.
- الموجات فوق الصوتية للمرارة والاثني عشر - في جميع مرضى GS تقريبًا ، لوحظت تغييرات في التركيب الكيميائي الحيوي للصفراء.
- خزعة الكبد - من الممكن حدوث تغييرات مرضية في العضو.
- اختبار الصيام - في وجود SF6 يؤدي سوء التغذية إلى زيادة البيليروبين في مصل الدم.
- اختبار مع الفينوباربيتال - يساعد استخدام الفينوباربيتال على خلفية SF على خفض مستوى البيليروبين غير المقترن.
- يؤدي اختبار حمض النيكوتينيك في SS إلى زيادة محتوى البيليروبين غير المقترن. يحدث نفس التفاعل مع إدخال الريفامبيسين.
قد يصف الطبيب دراسات إضافية وتشخيصًا تفاضليًا لـ SF مع فرط بيليروبين الدم.
 إن التكهن مواتٍ للغاية ، نظرًا لحقيقة أن مرض جيلبرت آمن نسبيًا ولا يتطلب علاجًا خاصًا (إنه ذو طبيعة محلية أكثر). أساس العلاج هو اتباع نظام غذائي عادي والعمل والراحة. أثناء التفاقم ، تحتاج إلى اتباع نظام غذائي رقم 5 (رفض الأطعمة الدهنية والمقلية ، والكحول) ، وتناول الفيتامينات والعوامل الصفراوية. قد يصف المعالج دورة من الأدوية الوقائية للكبد. من المهم أن تتذكر أنه لا يجب عليك اللجوء إلى العلاج الطبيعي الدافئ. يهدف علاج GS إلى استعادة المستوى الطبيعي لـ uridine diphosphate-glucuronyl transferase (إنزيم كبدي) واستقرار الحالة العامة للمريض.
إن التكهن مواتٍ للغاية ، نظرًا لحقيقة أن مرض جيلبرت آمن نسبيًا ولا يتطلب علاجًا خاصًا (إنه ذو طبيعة محلية أكثر). أساس العلاج هو اتباع نظام غذائي عادي والعمل والراحة. أثناء التفاقم ، تحتاج إلى اتباع نظام غذائي رقم 5 (رفض الأطعمة الدهنية والمقلية ، والكحول) ، وتناول الفيتامينات والعوامل الصفراوية. قد يصف المعالج دورة من الأدوية الوقائية للكبد. من المهم أن تتذكر أنه لا يجب عليك اللجوء إلى العلاج الطبيعي الدافئ. يهدف علاج GS إلى استعادة المستوى الطبيعي لـ uridine diphosphate-glucuronyl transferase (إنزيم كبدي) واستقرار الحالة العامة للمريض.
من الضروري التشاور مع المعالج وأخصائي أمراض الكبد عن الأدوية التي يمكن استخدامها على خلفية متلازمة جيلبرت ، وأي منها يجب التخلي عنه (على سبيل المثال ، يمكن أن تزيد الستيرويدات الابتنائية والقشرانيات السكرية والكافيين والباراسيتامول من ظهور اليرقان).
سرطان الأمعاء: الأعراض وعوامل الخطر
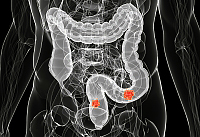 سرطان الأمعاء هو ورم خبيث يتشكل على الأغشية المخاطية في أجزاء مختلفة من الأمعاء. في أغلب الأحيان ، يمكن أن يتطور الورم من الاورام الحميدة ، ولكن لا تتحول جميعها إلى علم الأورام.
سرطان الأمعاء هو ورم خبيث يتشكل على الأغشية المخاطية في أجزاء مختلفة من الأمعاء. في أغلب الأحيان ، يمكن أن يتطور الورم من الاورام الحميدة ، ولكن لا تتحول جميعها إلى علم الأورام.
يمكن لسرطان الأمعاء أثناء تطوره أن يعطل الأداء الطبيعي للعضو ويؤدي إلى حدوث نزيف ، بما في ذلك تلك التي يمكن ملاحظتها في البراز. إذا لم يتم تشخيص سرطان الأمعاء في الوقت المناسب من خلال الأعراض ، فقد ينتشر إلى أعضاء أخرى ، مما يؤدي إلى تفاقم تشخيص العلاج بشكل كبير.
سرطان الأمعاء: عوامل الخطر
لا يذكر الطب الرسمي الأسباب الدقيقة لسرطان الأمعاء ، ولكنه يحدد عوامل الخطر الرئيسية التي تزيد من احتمالية الإصابة بأورام الأورام في الأمعاء:
- العمر: في أغلب الأحيان ، يعاني الأشخاص فوق سن الخمسين من سرطان الأمعاء ؛
- نمط حياة غير صحي: قلة النشاط البدني والوجبات السريعة والوزن الزائد وتعاطي الكحول والتدخين ؛
- : تعتبر العمليات الالتهابية في الأمعاء (مرض كرون ، والتهاب القولون التقرحي ، وما إلى ذلك) من الأمراض السرطانية التي تثير تطور الأورام ؛
- الوراثة: يزداد خطر الإصابة بالسرطان إذا أصيب أقرب الأقارب به أو بأمراض معوية أخرى.
سرطان الأمعاء: الأعراض
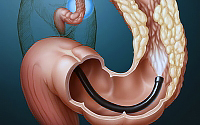 في المراحل المبكرة من سرطان الأمعاء ، قد لا تجذب الأعراض انتباه المريض. تغير في وتيرة حركات الأمعاء (في اتجاه التناقص أو الزيادة) ، وفقدان الوزن بدون سبب ، والتعب والضعف ، وفقر الدم ، وألم في فتحة الشرج - هذه هي العلامات الرئيسية التي تميز بداية عملية الأورام في الأمعاء.
في المراحل المبكرة من سرطان الأمعاء ، قد لا تجذب الأعراض انتباه المريض. تغير في وتيرة حركات الأمعاء (في اتجاه التناقص أو الزيادة) ، وفقدان الوزن بدون سبب ، والتعب والضعف ، وفقر الدم ، وألم في فتحة الشرج - هذه هي العلامات الرئيسية التي تميز بداية عملية الأورام في الأمعاء.
مع تطور المرض ، قد يحدث ألم بطني مستمر وانتفاخ وتدهور في الصحة العامة. يجب أن يكون ظهور أي من الأعراض سببًا وجيهًا لزيارة الطبيب فورًا.
إذا تم اكتشاف السرطان في المراحل المبكرة ، فإن تشخيص العلاج يكون متفائلًا تمامًا - حوالي 90 ٪ من المرضى يظلون على قيد الحياة ولم يعدوا يذهبون إلى الطبيب مع الشكاوى. ومع ذلك ، فإن معظم الناس لا تظهر عليهم الأعراض الأولى ، ويعتقدون أنها اضطراب أو عمليات البواسير ، مما يؤدي إلى ظهور المرض ، ومن ثم تنقذ الجراحة حوالي 60٪ فقط من المرضى.
لماذا هناك حاجة إلى أسنان إضافية؟
 الأسنان الزائدة ، أو فرط الأسنان ، هي واحدة من الحالات الشاذة المحددة وراثيًا في عدد الأسنان الشائعة جدًا في الوقت الحاضر. حوالي 2-3٪ من المرضى الذين يعانون من تشوهات الأسنان لديهم ، بالإضافة إلى 20 لبن أو 32 وحدة دائمة للأسنان. طبيعة هذه الحالة المرضية غير واضحة تمامًا ، يُعتقد أن حدوثها يرتبط بانتهاك وضع الأسنان ، أو بالأحرى ، بانتهاك آلية تقسيم صفيحة الأسنان ، مما يؤدي إلى تكوين المزيد من عدد جراثيم الأسنان.
الأسنان الزائدة ، أو فرط الأسنان ، هي واحدة من الحالات الشاذة المحددة وراثيًا في عدد الأسنان الشائعة جدًا في الوقت الحاضر. حوالي 2-3٪ من المرضى الذين يعانون من تشوهات الأسنان لديهم ، بالإضافة إلى 20 لبن أو 32 وحدة دائمة للأسنان. طبيعة هذه الحالة المرضية غير واضحة تمامًا ، يُعتقد أن حدوثها يرتبط بانتهاك وضع الأسنان ، أو بالأحرى ، بانتهاك آلية تقسيم صفيحة الأسنان ، مما يؤدي إلى تكوين المزيد من عدد جراثيم الأسنان.
أين تبحث عن أسنان إضافية؟
يمكن العثور على الأسنان الزائدة في الطفولة في انسداد اللبن ، ولكن في كثير من الأحيان يتم اكتشافها بعد تغيير الأسنان في الانسداد الدائم.
عادة ، تظهر الأسنان الزائدة بالقرب من القواطع العلوية الوسطى والأضراس والضواحك والأنياب ، وغالبًا ما تكون بين القواطع السفلية والضواحك والأنياب. يمكن أن تنمو على قوس الأسنان ، ويمكن أن تتواجد عشية تجويف الفم أو مباشرة في تجويف الفم في منطقة الحنك العلوي.
في الشكل ، قد تكون الأسنان الزائدة عن العدد مماثلة للوحدات الدائمة العادية للأسنان ، ولكنها غالبًا ما تكون على شكل دمعة أو على شكل مسمار. يمكن وضعها بشكل منفصل ، لحام بأسنان دائمة ، وتشكيل تكتلات أسنان كاملة وتشكيلات تشبه الأسنان.
في بعض الأحيان يتم "إخفاء" الأسنان الزائدة عن العين ، أي أنها متأثرة ، ثم يتم العثور عليها فقط أثناء فحص الأشعة السينية.
لماذا تعتبر الأسنان الزائدة خطرة؟
تتداخل الوحدات الزائدة مع تكوين الأسنان وتجعل من الصعب على الأسنان الدائمة انبثاقها. في حجم كبيرفي الفك ، الأسنان الزائدة لا تنتهك بنية الأسنان ، ومع وجود فك صغير ، فإنها بالتأكيد ستسبب شذوذًا في موضع الأسنان الكاملة ، الأمر الذي له عواقب جمالية ووظيفية ونفسية سلبية على الإنسان في كثير من الأحيان.
غالبًا ما يعاني الأطفال الذين يعانون من تشوهات في نمو الأسنان من انخفاض الشهية ، فهم يمضغون الطعام بشكل أبطأ ، وغالبًا ما يعانون من ضعف في البلع ، كل هذا يتسبب في تطور أمراض الجهاز الهضمي.
الترتيب الوثيق للأسنان ، ووضعها غير الصحيح يجعل من الصعب عمليات التنظيف الذاتي للأسنان وتنفيذ إجراءات النظافة. يخلق الظروف المواتيةلتطور تسوس الأسنان والتهاب اللثة والتهاب دواعم الأسنان وأمراض اللثة مما يؤدي إلى تدمير الأنسجة الصلبة للأسنان وفقدانها.
أصحاب الأسنان الزائدة غالبًا ما يصابون بالثغرات. تتسبب اضطرابات النطق والقصور التجميلي في السخرية من الطفل ، وتجعله في شخصية غير تواصلية ومنطوية ، وغالبًا ما تؤثر على نموه العقلي.
تشخيص فرط الأسنان
في معظم الحالات ، توجد الأسنان الزائدة عن العدد أثناء بزوغ الأسنان الأمامية. لتوضيح عددهم وموقعهم ، فإن التشخيص بالأشعة السينية ضروري ، لكن التصوير الشعاعي البسيط لا يكفي ، حيث يتم تثبيت ظلال الأسنان الزائدة على ملامح العناصر الكاملة للأسنان.
للتشخيص الدقيق لفرط الأسنان ، يتم استخدام الأشعة السينية داخل الفم مع صور في إسقاطات مختلفة. مع الاحتفاظ المتعدد للأسنان الزائدة عن العدد ، يوفر تقويم العظام معلومات مفيدة حول الوضع النسبي للأسنان الزائدة والدائمة.
ماذا تفعل بالأسنان الزائدة؟
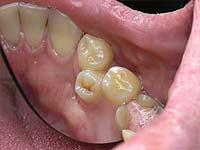 نهج علاج فرط الأسنان متمايز ، يعتمد العلاج على موقع الأسنان الإضافية. بشكل عام ، يجب إزالة الأسنان الزائدة في أسرع وقت ممكن ، خاصة إذا كانت تنتهك تشريح الأسنان وتسبب ألمًا للمريض. مع التخلص من الأسنان الزائدة في مرحلة الطفولة ، غالبًا ما يتم استعادة الشكل الطبيعي للأسنان بسبب آليات التنظيم الذاتي للجسم ، ولكن إذا فاتك الوقت ، فلا يمكنك الاستغناء عن العلاج التقويمي اللاحق.
نهج علاج فرط الأسنان متمايز ، يعتمد العلاج على موقع الأسنان الإضافية. بشكل عام ، يجب إزالة الأسنان الزائدة في أسرع وقت ممكن ، خاصة إذا كانت تنتهك تشريح الأسنان وتسبب ألمًا للمريض. مع التخلص من الأسنان الزائدة في مرحلة الطفولة ، غالبًا ما يتم استعادة الشكل الطبيعي للأسنان بسبب آليات التنظيم الذاتي للجسم ، ولكن إذا فاتك الوقت ، فلا يمكنك الاستغناء عن العلاج التقويمي اللاحق.
في بعض الأحيان ، من أجل الحفاظ على وظائف الأسنان ، على العكس من ذلك ، يتم التضحية بالأسنان الدائمة البائسة ، مع الحفاظ على سن زائدة تشريحيا مكتملة الموقع بشكل مفيد.
إذا نمت السن الزائد بدلاً من سن دائم غير مقطوع ، يتم تحديد درجة فائدته أولاً. في حالة ثبات السن الزائد ، وله جذر متطور وشكل صحيح تشريحيًا إلى حد ما ، و "المالك الشرعي للمكان" الذي لم ينفجر لا يمكن الدفاع عنه وغير واعد ، يتم إعطاء الأفضلية لـ "الغازي".
تؤدي إزالة الأسنان الزائدة المحطمة المغمورة في أنسجة الفك إلى بعض الصعوبات المرتبطة بعمقها وشكلها غير المنتظم وقربها من جذور وأساسيات الأسنان الدائمة. ومع ذلك ، فإن النهج التشغيلي العقلاني للأسنان الزائدة ، مع مراعاة نتائج فحص الأشعة السينية ، يسمح لنا بحل هذه المشكلات بنجاح.
في الأطفال ، يتم إزالة الأسنان الزائدة عن الحد تحت التخدير العام أو الموضعي ، على خلفية عمل الأدوية المهدئة ، عند البالغين ، يكون التخدير الموضعي كافياً.
بورفيريا - "مصاص دماء" قائم على العلم
مصاصي الدماء هي ثقافة فرعية حديثة توحد الشباب الذين يعتبرون أنفسهم مصاصي دماء. في الأساس ، يقتصر الاهتمام على دراسة موضوعات مصاصي الدماء في الفن والتقليد مظهرالشخصيات المفضلة ، من غير المرجح أن يفكر الشباب في تاريخ أصل صور مصاصي الدماء.
حالات مصاص دماء "غير علمي"
ولدت حركة مصاصي الدماء عام 1970 بفضل محبي أعمال آن رايس ، مؤلفة الرواية الشهيرة مقابلة مع مصاص الدماء. في الوقت نفسه ، فإن موضوع مصاص الدماء له جذوره في الماضي البعيد وينعكس في الفولكلور للعديد من الشعوب.
تطورت صورة مصاص الدماء في الفن على مر السنين ، لكنها انعكست بشكل أوضح في الرواية القوطية دراكولا للكاتب الأيرلندي برام ستوكر ، التي نُشرت في عام 1897 ، والأعمال اللاحقة تدين بوجودها لهذا الإبداع الخالد.
ما هي العلامات الأدبية لمصاصي الدماء؟
 في أغلب الأحيان ، يتم تصوير مصاصي الدماء على أنهم أفراد أذكياء وأنيقون وغامضون ومثيرون يعيشون أسلوب حياة منعزل.
في أغلب الأحيان ، يتم تصوير مصاصي الدماء على أنهم أفراد أذكياء وأنيقون وغامضون ومثيرون يعيشون أسلوب حياة منعزل.
إنهم بحاجة إلى الدم من أجل الحفاظ على التمثيل الغذائي وعدم الموت. بحسب الأساطير:
- يخاف مصاصو الدماء من أشعة الشمس ، ويتم حمايتهم بالملابس الداكنة ، ولا يخرجون إلا تحت جنح الليل ويعودون قبل الفجر ؛
- يقتل ضوء النهار مصاص الدماء ويقلل من قوته ؛
- يتجنبون حفلات العشاء والعشاء ، فالغذاء البشري غريب عليهم ؛
- تكون شاحبة ، والجلد رقيق وضعيف ، وبارد عند اللمس ؛
- لم تتغير - الأنياب واللثة الأرجوانية.
- عيون مصاص الدماء محاطة بضباب من الرموش الرقيقة ، والبيض محمر ، والتلاميذ غائمتان ؛
- إنهم يتسمون بالقلق ، والشك ، والعدوانية ، ويذهبون هائجون في لحظة الرغبة الشديدة في الدم ، ويمكن أن يتحولوا إلى وحوش بالمعنى الجسدي والنفسي.
البورفيريا
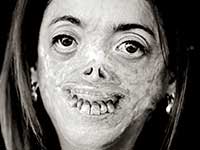 عدم امتلاك أي معرفة في مجال الطب ، واستلهامًا من الأساطير الأيرلندية حول مصاصي الدماء ، ومن أساطير شعوب ترانسيلفانيا ، والأوصاف التاريخية لحياة فلاد المخوزق ، النموذج الأولي لدراكولا ، وصف ستوكر ، دون الشك في ذلك ، المعاناة لشخص مصاب بمرض خطير من البورفيريا.
عدم امتلاك أي معرفة في مجال الطب ، واستلهامًا من الأساطير الأيرلندية حول مصاصي الدماء ، ومن أساطير شعوب ترانسيلفانيا ، والأوصاف التاريخية لحياة فلاد المخوزق ، النموذج الأولي لدراكولا ، وصف ستوكر ، دون الشك في ذلك ، المعاناة لشخص مصاب بمرض خطير من البورفيريا.
البورفيريا ، خلاف ذلك - مرض أرجواني ، مجموعة من الأمراض المرتبطة بضعف التمثيل الغذائي للبورفيرين ، صبغة حمراء زاهية. يعتمد علم الأمراض على انتهاكات تخليق الهيم - مركب البورفيرين بالحديد ، وهو أساس كريات الدم الحمراء البشرية. يؤدي الفشل في نظام تكوين الهيم إلى فقر الدم ، وتراكم المنتجات الأيضية الوسيطة في الجسم ، والتي لها تأثير سام على الأعضاء والأنظمة ، مما يتسبب في ظهور الأعراض النموذجية لمرض "مصاص الدماء".
تكمن أسباب تطور البورفيريا المستوى الجيني, المرض وراثي. إن احتمال نقل جين البورفيريا مرتفع للغاية ، فالوالد المريض "يعطي" الجين المعيب للطفل في 50٪ من الحالات ، بغض النظر عن الجنس ، ولكن في 20٪ فقط من الحالات تظهر الصورة السريرية للمرض. يحتاج إلى عمل لإظهاره. عوامل استفزازية: بعض الأدوية ، والالتهابات ، والتغيرات الهرمونية ، وبعض الأطعمة والكحول - لم يكن من أجل لا شيء أن مصاصي الدماء الأسطوريين تجنبوا الأعياد البشرية.
أعراض البورفيريا
حيث أن الأمراض أكثر شيوعًا بين الرجال وتظهر في فصلي الربيع والصيف. الأعراض النموذجية للبورفيريا هي البول الأحمر البني، والذي يرجع إلى وجود البورفيرينوجين ناقص الأكسدة ، والذي يتحول إلى بورفيرين أرجواني في الضوء.
البورفيريا الحادةيبدو ألم حادفي البطن ، أسفل الظهر ، في الأطراف ، عدم انتظام دقات القلب ، ارتفاع ضغط الدم ، القيء ، ضعف العضلات ، التحريض النفسي ، الهلوسة ، الهذيان ، النوبات الصرعية والأعراض الأخرى التي تتطور نتيجة التسمم الحاد في الجسم ، التي تكونت من المنتجات من استقلاب البورفيرين وانتشار الضرر على الأطراف والمركز الجهاز العصبي.
البورفيريا لها مظاهر خارجية نموذجية للغاية.
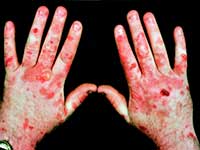
يمكن للمرء أن يتخيل رد فعل الناس في العصور الوسطى عند مواجهة مريض مصاب بالبورفيريا. مثل هذا المشهد ترك بصمة في الذاكرة ، مطبوعة في الأساطير ، متضخمة بالتفاصيل الأسطورية.
البورفيريا مرض خطير يتم علاجه بشكل رئيسي عن طريق تسريب الهيم. بالإضافة إلى ذلك ، يتم وصف عوامل الأعراض ، فصادة البلازما ، والدم ضروري للمرضى من أجل التعامل مع التسمم والعيش فقط.
مشكلة حقيقية للرجل: العقم وأسبابه
 يقولون إن كل رجل في حياته يجب أن يزرع شجرة ويبني منزلاً ويربي ولداً. وإذا تعامل كل فرد من أفراد الجنس الأقوى تقريبًا مع المهمتين الأوليين ، إذا رغبت في ذلك ، فعندئذ ، للأسف ، عند حل آخرهما ، يواجه ما يصل إلى 8٪ من الرجال على وجه الأرض مشاكل خطيرة. وهذا الرقم لا يأخذ في الاعتبار أولئك الذين تعاملوا مع العقم واختاروا عدم مشاركة مشكلتهم مع الأطباء.
يقولون إن كل رجل في حياته يجب أن يزرع شجرة ويبني منزلاً ويربي ولداً. وإذا تعامل كل فرد من أفراد الجنس الأقوى تقريبًا مع المهمتين الأوليين ، إذا رغبت في ذلك ، فعندئذ ، للأسف ، عند حل آخرهما ، يواجه ما يصل إلى 8٪ من الرجال على وجه الأرض مشاكل خطيرة. وهذا الرقم لا يأخذ في الاعتبار أولئك الذين تعاملوا مع العقم واختاروا عدم مشاركة مشكلتهم مع الأطباء.
العقم عند الرجل هو سبب عقم الزوجين
لا يقتصر العقم على غياب الأطفال فقط ، بل يتم تشخيصه إذا كان الزوجان النشطان جنسياً دون استخدام وسائل منع الحمل يواجهان صعوبة في الحمل خلال العام. دعنا نوضح مفهوم "الحياة الجنسية النشطة" قليلاً ، فهو بالنسبة لشخص ما هو الجنس اليومي ، والآخر - مرتين في الشهر. في حالة العقم ، تعني كلمة "نشط" مرة واحدة على الأقل في الأسبوع.
وفقًا للإحصاءات ، يقع اللوم في 40٪ من الحالات التي لا ينجب فيها الزوجان أطفالًا. يمكن أن تكون أسباب ذلك مختلفة ، للوهلة الأولى غير ضارة ويمكن التغلب عليها بسهولة ، أو كبيرة لدرجة أنه من المستحيل القضاء عليها.
يمكن أن يكون العقم عند الذكور ، وكذلك العقم عند النساء ، مطلقًا ونسبيًا. سيضطر الرجل إلى تحمل العقم المطلق إذا تمت إزالة الغدد المنوية. من المحتمل أن تكون المتغيرات الأخرى للعقم نسبية ، وتتطلب تشخيصات متعمقة للعثور على السبب وحل المشكلة.
متغيرات العقم عند الذكور: الأسباب والأشكال
 عملية تكوين الحيوانات المنوية الكاملة معقدة للغاية ويتم التحكم فيها من خلال الجزء المنتج للهرمونات في الدماغ - نظام الغدة النخامية - الوطاء. تنضج الخلايا الجنسية التي تتكون في الخصية في طريقها إلى الحويصلات المنوية ، لكن عدم وجود مشاكل في هذا الجزء من الإنتاج لا يضمن نجاح الإخصاب. تتأثر القدرة الإنجابية للرجل بحالة جميع الأعضاء الذكرية والكائن الحي ككل.
عملية تكوين الحيوانات المنوية الكاملة معقدة للغاية ويتم التحكم فيها من خلال الجزء المنتج للهرمونات في الدماغ - نظام الغدة النخامية - الوطاء. تنضج الخلايا الجنسية التي تتكون في الخصية في طريقها إلى الحويصلات المنوية ، لكن عدم وجود مشاكل في هذا الجزء من الإنتاج لا يضمن نجاح الإخصاب. تتأثر القدرة الإنجابية للرجل بحالة جميع الأعضاء الذكرية والكائن الحي ككل.
ما الذي يسبب هذه المشكلة؟
- اضطرابات الغدة النخامية التي تحدث في الدماغ على مستوى التنظيم الهرموني لتكوين الحيوانات المنوية.
- الخصية ، المرتبطة بالخصيتين.
- ما بعد الخصية ، وتنشأ على طول حركة الحيوانات المنوية من الخصية إلى مخرج مجرى البول.
- مناعي مع وجود الأجسام المضادة للحيوانات المنوية التي تقتل الحيوانات المنوية.
- اضطرابات القذف أي مشكلة القذف.
- الاضطرابات الجنسية التي تعطل "توصيل" الحيوانات المنوية إلى الأعضاء التناسلية للمرأة ، مثل ضعف الانتصاب وانخفاض الرغبة الجنسية.
تتأثر قدرة الرجل على الإنجاب سلبًا بما يلي:
- الوضع البيئي غير المواتي
- ضغط؛
- تعاطي المخدرات بجرعات عالية.
- التدخين؛
- تناول الكحول؛
- تعاطي المخدرات.
العقم الخصوي
يحدث عقم الخصية مع أمراض الخصية والقصور الذاتي.
- دوالي الخصية في كيس الصفن والحبل المنوي (دوالي الخصية).
- الخصية الخفية ، ثنائية لم يتم القضاء عليها أو القضاء عليها في وقت متأخر جدًا ، عندما يكون النسيج المنوي للخصيتين قد ضمر بالفعل.
- التواء الخصية ، مما يؤدي إلى انتهاك حاد لوظيفتها.
- التهاب الخصية ، أو التهاب الخصية ، مما يؤدي إلى موت الخلايا السلفية للحيوانات المنوية.
- قصور الغدد التناسلية مفرط الغدد التناسلية هو تخلف في الخصيتين.
- الأسباب الوراثية التي تسبب انتهاكًا لتكوين الجهاز التناسلي وتكوين الخصيتين.
- مقاومة الأندروجين ، عندما تظل الظهارة المولدة للحيوانات المنوية في الخصيتين صماء أمام الإشارات الهرمونية التي تعلن عن تكوين الخلايا الجرثومية.
- فقد النطاف غير الانسدادي ، وهي حالة لا يضعف فيها إنتاج الحيوانات المنوية ، وحركتها على طول الأسهر ليست صعبة ، ولكن "لا توجد عينة حية واحدة" قادرة على استمرار الجنس عند المخرج.
أسباب العقم بعد الخصية
تؤدي هذه الأسباب إلى ضعف نضج الحيوانات المنوية ، وموتها ، وإضعاف قدرتها على التخصيب ، كما تمنع حركة الحيوانات المنوية عبر الأسهر.
- التهابات الأعضاء التناسلية ، خاصة تلك طويلة الأمد والكامنة ، على سبيل المثال ، الكلاميديا ، داء المشعرات ، داء المفطورات ، داء البول ، عدوى الفيروس المضخم للخلايا ، الهربس.
- التهاب غير محدد في البروستاتا والإحليل والحويصلات المنوية.
- غياب البربخ ، الذي "تنضج" فيه الحيوانات المنوية ، والقناة التي تخرج من خلالها الخصية.
- انسداد الأسهر أو إزالتها.
أخيرًا ، هناك خيارات عندما لا يمكن تحديد أسباب تدهور الوظيفة الإنجابية ، ما يسمى بالعقم مجهول السبب. في أي حال ، للحصول على تشخيص نهائي ، تحتاج إلى الاتصال بأخصائي المسالك البولية والخضوع لفحص كامل.
يشير داء السكري من النوع الأول ، أي أنه يوجد عند الأطفال ، إلى الأمراض ، والاستعداد للإصابة بالوراثة. إنه الاستعداد ، ولكن ليس المرض نفسه ، الذي يتطور فقط عندما يكون استفزازيًا خارجيًا و العوامل الداخلية. لم يتم تحديد جميع أسباب داء السكري حتى الآن ، ولكن يُعتقد أن العدوى أو الإجهاد غالبًا ما يصبحان نقطة البداية للمرض.

يبدو أنه إذا كان الوالدان أو أقارب الطفل الآخرون على دراية بأسباب وعلامات مرض السكري ، لأنهم يعانون منه ، فلا ينبغي أن تكون هناك أي صعوبات في الكشف المبكر عن المرض لدى الطفل. ومع ذلك ، لسوء الحظ ، يوجد المرض في أغلب الأحيان
المرحلة المتأخرة ، عندما يكون الطفل في حالة خطيرة بالفعل ويحتاج بشدة إلى إجراءات طبية مكثفة. من المحزن أن نقول ، ولكن في هذه المرحلة ، يصعب علاج مرض السكري ويسبب مضاعفات خطيرة. الوضع معقد بسبب حقيقة أن مرض السكري في مرحلة الطفولة له فترة كامنة قصيرة جدًا ، مما يعني أنه لا يوجد وقت للتفكير الطويل.
يجب أن يعرف كل فرد من أفراد الأسرة البالغين بوضوح العلامات الأولى لمرض السكري لدى الأطفال وأن يكون قادرًا على تقييم المؤشرات الصحية الأكثر أهمية من حيث التشخيص المبكر للمرض.
ما هي علامات مرض السكري التي يجب على الآباء الانتباه لها؟
- تغير الشهية:
- ظهور شغف غير طبيعي بالحلويات للطفل ؛
- الطموح في تناول الطعام في كثير من الأحيان ، أي أن الطفل ، بسبب الشعور القوي بالجوع ، لا يمكنه تحمل فترات الراحة التقليدية 3-4 ساعات بين الوجبات ؛
- الضعف والنعاس 1.5-2 ساعة بعد الأكل.
بالطبع ، يحب معظم الأطفال الحلويات ، ويميل الكثيرون إلى النوم بعد الأكل ، ولكن في حالة الاستعداد الوراثي لمرض السكري من النوع الأول ، قد تكون هذه العادات هي أولى علامات المرض ، ووجودهم المنعزل قد يشير إلى أن المرض قد أصابهم. لم تذهب بعيدًا بعد ، مما يعني أنه سيكون من الأسهل علاجها. - يفقد الطفل وزنه ، على الرغم من شهيته الطبيعية وحتى المتزايدة.
لا داعي لأن نعزو فقدان الوزن إلى حقيقة أن الطفل ينمو بسرعة ، فمن الأفضل فحصه والتأكد من أن التغيير في وزن الجسم ناتج عن زيادة النشاط البدني وزيادة احتياجات الطفل وليس بحقيقة أن جسده يحاول بكل قوته محاربة المرض. على الرغم من احتواء الدم على كمية زائدة من الجلوكوز ، فإن خلايا جسم الطفل تعاني من الجوع الشديد ، لأن البنكرياس المصاب بداء السكري لا يصنع عمليًا الأنسولين ، وهو أمر ضروري لامتصاص الجلوكوز من الأنسجة. - سرعان ما يتعب الطفل ، ويصبح خاملًا ، وينام.
في حالة عدم وجود حمى وسعال وغيرها من علامات البرد ، يجب أن تنبهك هذه الأعراض فيما يتعلق بمرض السكري. - يبدأ الطفل في الشرب والتبول أكثر ، على الرغم من أن أطباء السكري يتعرفون على هذه الأعراض في وقت متأخر.
مثل الملح ، يجذب السكر السائل إلى نفسه ، والجسم ، في محاولة "لتخفيف" السكر ، يحتاج إلى الماء ، مما يشير إلى ذلك بالعطش. عاجلاً أم آجلاً ، مع مرض السكري ، تتوقف الكلى عن الاحتفاظ بالجلوكوز في الجسم ، ويبدأ في إفرازه في البول ، مما يؤدي بدوره إلى زيادة إدرار البول. يبدأ الأطفال المصابون بداء السكري في الاستيقاظ ليلاً للذهاب إلى المرحاض ، وفي بعض الأحيان يقومون بتبليل الفراش. - بقع البول على القصرية ، في المرحاض ، على الحفاضات تصبح لزجة.
هذه ظاهرة فيزيائية بحتة ، محلول السكر ، لأسباب واضحة ، يترك وراءه بقعًا لزجة بعد تبخر الماء. ستلاحظ الأم اليقظة دائمًا هذه الأعراض.
غثيان، قيء ، آلام في البطن ، جفاف وحكة في الجلد ، علامات التهاب الجلد العصبي ، داء جلدي مستمر ، تقيح الجلد ، ضعف البصر - هذه أعراض وعواقب متأخرة لمرض السكري ، وهي علامة على أن المرض قد اكتسب قوة بالفعل وسيكون من الصعب للغاية لوقف تطورها. ولكن إذا لجأ الوالدان إلى اختصاصي الغدد الصماء عند ظهور الإشارات الأولى لمرض السكري ، يمكن اكتشاف المرض في مرحلة مبكرة ، حتى قبل أن يتعطل البنكرياس ويزيد مستوى السكر في الدم. لن يضيع الوقت وسيكون الأطباء قادرين على توفير قوة الأطفال لمحاربة المرض ولحياة كاملة.
انتباه! يجب أن يكون جميع البالغين على دراية بوجود مرض السكري عند الطفل:المعلمين والمعلمين والجيران والأصدقاء على اتصال معه. أولاً ، سيتجنب ذلك الأخطاء في النظام الغذائي للطفل ، وثانيًا ، في حالة حدوث تدهور مفاجئ في صحته ، اصطحبه إلى قسم الغدد الصماء المتخصص ، وليس على سبيل المثال ، مستشفى الأمراض المعدية أو الجراحة.
من بين الأمراض الوراثية التي تتطور نتيجة الطفرات ، يتم تمييز ثلاث مجموعات فرعية تقليديًا:
- أمراض وراثية أحادية الجين
- أمراض وراثية متعددة الجينات
- الانحرافات الصبغية
من الأمراض الوراثية يجب التمييز بين الأمراض الخلقية التي تسببها أضرار داخل الرحم ناتجة ، على سبيل المثال ، عن طريق العدوى (الزهري أو داء المقوسات) أو التعرض لعوامل ضارة أخرى على الجنين أثناء الحمل.
لا تظهر العديد من الأمراض المحددة وراثيًا بعد الولادة مباشرة ، ولكن بعد مرور بعض الوقت ، وأحيانًا لفترة طويلة جدًا.
أمراض وراثية أحادية المنشأ
يتم توريث الأمراض أحادية الجين وفقًا لقوانين علم الوراثة الكلاسيكي المندلي. وفقًا لذلك ، بالنسبة لهم ، يكشف البحث في علم الأنساب عن نوع واحد من ثلاثة أنواع من الميراث: الوراثة الجسدية السائدة ، والوراثة الجسدية المتنحية ، والميراث المرتبط بالجنس.
هذه هي أوسع مجموعة من الأمراض الوراثية. حاليًا ، تم وصف أكثر من 4000 نوع من الأمراض الوراثية أحادية الجين ، والغالبية العظمى منها نادرة جدًا (على سبيل المثال ، معدل حدوث فقر الدم المنجلي هو 1/6000).
مجموعة واسعة من الأمراض أحادية الجين تشكل اضطرابات استقلابية وراثية ، يرتبط حدوثها بطفرة في الجينات التي تتحكم في تخليق الإنزيمات وتسبب نقصها أو عيبها الهيكلي - اعتلال الخميرة.
أمراض وراثية متعددة الجينات
يصعب توريث الأمراض متعددة الجينات. بالنسبة لهم ، لا يمكن البت في مسألة الميراث على أساس قوانين مندل. في السابق ، كانت هذه الأمراض الوراثية توصف بأنها أمراض ذات استعداد وراثي. تشمل هذه الأمراض أمراضًا مثل السرطان والسكري وانفصام الشخصية والصرع وأمراض القلب التاجية وارتفاع ضغط الدم وغيرها الكثير.
الانحرافات الصبغية
تنجم أمراض الكروموسومات عن انتهاك جسيم للجهاز الوراثي - تغيير في عدد وهيكل الكروموسومات. السبب النموذجي ، على وجه الخصوص ، هو تسمم الكحول للوالدين عند الحمل ("الأطفال في حالة سكر"). وتشمل هذه متلازمة داون ، وكلينفيلتر ، وشيريشيفسكي - تيرنر ، وإدواردز ، و "صرخة القطة" وغيرها.
تشخيص وعلاج الأمراض الوراثية
في الآونة الأخيرة ، كان هناك رأي مفاده أن التكرار المرتفع نسبيًا للأمراض الوراثية يرجع إلى بعض مزايا "الطفرات" فيما يتعلق بالعوامل الانتقاء الطبيعيأو مع "القابلية للإصابة بالمرض".
يشمل علاج الأمراض الوراثية علاج الأعراض والعلاج الجيني.
علاج الأعراض
تتميز الأمراض الوراثية بمظاهر عرضية مختلفة ، وعلاجها عرضي إلى حد كبير. يتم تصحيح بعض اضطرابات التمثيل الغذائي عن طريق تعيين أنظمة غذائية خاصة تهدف إلى تقليل المواد السامة في الجسم ، والتي يرجع تراكمها إلى طفرات في جينات معينة. على سبيل المثال ، مع بيلة الفينيل كيتون ، يتم وصف نظام غذائي خالٍ من الألانين.
للتخفيف من أعراض الأمراض الوراثية المصاحبة لخلل في بروتين معين ، يتم إعطاء شكله الوظيفي عن طريق الوريد ، مما لا يسبب استجابة مناعية. يتم استخدام هذا العلاج البديل في علاج الهيموفيليا ونقص المناعة المشترك الشديد وما إلى ذلك. في بعض الأحيان يتم زرع نخاع العظام والأعضاء الأخرى للتعويض عن وظائف معينة مفقودة. العلاج الحالي ، للأسف ، في الغالبية العظمى من الحالات غير فعال للغاية.
العلاج الجيني
العلاج الجيني هو طريقة جديدة وفعالة تهدف إلى تدمير السبب الجيني لمرض وراثي. جوهر طريقة العلاج الجيني هو إدخال الجينات الطبيعية في الخلايا المعيبة.
مفهوم العلاج الجيني هو أن الطريقة الأكثر جذرية لمكافحة أنواع مختلفة من الأمراض التي تسببها التغيرات في المحتوى الجيني للخلايا يجب أن تكون علاجًا يهدف مباشرة إلى تصحيح أو تدمير السبب الجيني للمرض ، وليس عواقبه.
يرجع ذلك إلى حقيقة أن العلاج الجيني هو اتجاه جديد علم الوراثة الطبية، والأمراض التي تحاول معالجتها بهذه الطريقة متنوعة للغاية ، وقد تم إنشاء العديد من الأساليب الأصلية لهذه المشكلة. حاليًا ، تهدف الأبحاث في العلاج الجيني بشكل أساسي إلى تصحيح العيوب الوراثية في الخلايا الجسدية بدلاً من الخلايا الجرثومية ، والتي ترتبط بمشاكل تقنية بحتة ، وكذلك لأسباب تتعلق بالسلامة.
بحسب مقال "الأمراض الوراثية"
لا يمكن أن ترث فقط علامات خارجيةولكن أيضًا الأمراض. نتيجة لذلك ، يؤدي الفشل في جينات الأجداد إلى عواقب في النسل. سنتحدث عن الأمراض الوراثية السبعة الأكثر شيوعًا.
تنتقل الخصائص الوراثية إلى أحفاد الأسلاف في شكل جينات مدمجة في كتل تسمى الكروموسومات. تحتوي جميع خلايا الجسم ، باستثناء الخلايا الجنسية ، على مجموعة مزدوجة من الكروموسومات ، نصفها من الأم ، والجزء الثاني من الأب. الأمراض التي تسببها عيوب معينة في الجينات وراثية.
قصر النظر
أو قصر النظر. مرض محدد وراثيا ، جوهره أن الصورة لا تتشكل على شبكية العين ، ولكن أمامها. يعتبر السبب الأكثر شيوعًا لهذه الظاهرة هو تضخم مقلة العين. كقاعدة عامة ، يتطور قصر النظر خلال فترة المراهقة. في الوقت نفسه ، يرى الشخص قريبًا جيدًا ، لكنه يرى بشكل ضعيف من مسافة بعيدة.
إذا كان كلا الوالدين مصابين بقصر النظر ، فإن خطر الإصابة بقصر النظر لدى أطفالهم يزيد عن 50٪. إذا كان كلا الوالدين يتمتعان برؤية طبيعية ، فإن احتمال الإصابة بقصر النظر لا يزيد عن 10٪.
البحث عن قصر النظر ، والموظفين الاستراليين جامعة وطنيةفي كانبيرا خلص إلى أن قصر النظر متأصل في 30 ٪ من الممثلين العرق القوقازيويؤثر على ما يصل إلى 80 ٪ من السكان الأصليين في آسيا ، بما في ذلك سكان الصين واليابان وكوريا الجنوبية ، وما إلى ذلك. وبعد جمع البيانات من أكثر من 45 ألف شخص ، حدد العلماء 24 جينًا مرتبطًا بقصر النظر ، وأكدوا أيضًا ارتباطهم باثنين الجينات التي تم إنشاؤها مسبقًا. كل هذه الجينات مسؤولة عن نمو العين وهيكلها ، والإشارات في أنسجة العين.
متلازمة داون
المتلازمة ، التي سميت على اسم الطبيب الإنجليزي جون داون ، الذي وصفها لأول مرة في عام 1866 ، هي شكل من أشكال الطفرات الصبغية. متلازمة داون تصيب جميع الأجناس.
المرض ناتج عن حقيقة أنه لا توجد نسختان ، بل ثلاث نسخ من الكروموسوم الحادي والعشرين في الخلايا. يسمي علماء الوراثة هذا التثلث الصبغي. في معظم الحالات ، ينتقل الكروموسوم الإضافي إلى الطفل من الأم. من المقبول عمومًا أن خطر إنجاب طفل مصاب بمتلازمة داون يعتمد على عمر الأم. ومع ذلك ، نظرًا لحقيقة أنهم ، بشكل عام ، يولدون في الغالب في سن الشباب ، فإن 80 ٪ من جميع الأطفال المصابين بمتلازمة داون يولدون لنساء دون سن 30 عامًا.
على عكس الجينات ، فإن تشوهات الكروموسومات هي حالات فشل عشوائية. وفي الأسرة يمكن أن يكون هناك شخص واحد فقط يعاني من هذا المرض. ولكن حتى هنا توجد استثناءات: في 3-5 ٪ من الحالات ، هناك أكثر ندرة - أشكال الانتقال من متلازمة داون ، عندما يكون لدى الطفل بنية أكثر تعقيدًا من مجموعة الكروموسومات. يمكن تكرار نوع مماثل من المرض في عدة أجيال من نفس العائلة.
وفقًا لمؤسسة Downside Up الخيرية ، يولد حوالي 2500 طفل مصاب بمتلازمة داون في روسيا كل عام.
متلازمة كلاينفلتر
اضطراب كروموسومي آخر. تقريبًا لكل 500 ولد حديث الولادة ، هناك طفل مصاب بهذه الحالة المرضية. تظهر متلازمة كلاينفيلتر عادة بعد سن البلوغ. الرجال الذين يعانون من هذه المتلازمة يعانون من العقم. بالإضافة إلى ذلك ، فهي تتميز بالتثدي - زيادة في الغدة الثديية مع تضخم في الغدد والأنسجة الدهنية.
حصلت المتلازمة على اسمها تكريما للطبيب الأمريكي هاري كلاينفيلتر ، الذي وصف الصورة السريرية لأول مرة لعلم الأمراض في عام 1942. بالاشتراك مع اختصاصي الغدد الصماء فولر أولبرايت ، وجد أنه إذا كان لدى النساء عادةً زوج من الكروموسومات الجنسية XX ، وكان لدى الرجال XY ، فعند هذه المتلازمة ، يكون لدى الرجال من واحد إلى ثلاثة كروموسومات X إضافية.
عمى الألوان
أو عمى الألوان. إنه وراثي ، نادرًا ما يتم الحصول عليه. يتم التعبير عنها في عدم القدرة على تمييز لون واحد أو أكثر.
يرتبط عمى الألوان بالكروموسوم X وينتقل من الأم ، صاحبة الجين "المكسور" ، إلى ابنها. وعليه ، فإن ما يصل إلى 8٪ من الرجال ولا يزيد عن 0.4٪ من النساء يعانون من عمى الألوان. والحقيقة هي أنه عند الرجال ، لا يتم تعويض "الزواج" في كروموسوم X واحد ، لأنهم لا يمتلكون كروموسوم X ثانٍ ، على عكس النساء.
الهيموفيليا
مرض آخر يورثه الأبناء من الأمهات. قصة أحفاد الملكة الإنجليزية فيكتوريا من سلالة وندسور معروفة على نطاق واسع. لم تعاني هي ولا والديها من هذا المرض الخطير المرتبط بضعف تخثر الدم. من المفترض أن الطفرة الجينية حدثت تلقائيًا بسبب حقيقة أن والد فيكتوريا في وقت حملها كان بالفعل يبلغ من العمر 52 عامًا.
ورث الأطفال الجين "القاتل" من فيكتوريا. توفي ابنها ليوبولد بسبب مرض الهيموفيليا عن عمر يناهز 30 عامًا ، وحملت اثنتان من بناتها الخمس ، أليس وبياتريس ، الجين المشؤوم. أحد أشهر أحفاد فيكتوريا الذين عانوا من مرض الهيموفيليا هو ابن حفيدتها ، تساريفيتش أليكسي ، الابن الوحيد لآخر إمبراطور روسي نيكولاس الثاني.
تليّف كيسي
مرض وراثي يتجلى في تمزق الغدد المفرزة الخارجية. يتميز بزيادة التعرق وإفراز المخاط الذي يتراكم في الجسم ويمنع نمو الطفل ، والأهم من ذلك أنه يمنع عمل الرئتين بشكل كامل. الموت المحتمل بسبب فشل الجهاز التنفسي.
وفقًا للفرع الروسي لشركة المواد الكيميائية والصيدلانية الأمريكية Abbott ، يبلغ متوسط العمر المتوقع لمرضى التليف الكيسي في الدول الأوروبية 40 عامًا ، في كندا والولايات المتحدة - 48 عامًا ، في روسيا - 30 عامًا. ومن الأمثلة الشهيرة المغني الفرنسي غريغوري ليمارشال ، الذي توفي عن عمر يناهز 23 عامًا. يُفترض أن فريدريك شوبان عانى أيضًا من التليف الكيسي ، وتوفي نتيجة لفشل الرئة عن عمر يناهز 39 عامًا.
مرض ورد ذكره في البرديات المصرية القديمة. من الأعراض المميزة للصداع النصفي نوبات صداع شديدة عرضية أو منتظمة في جانب واحد من الرأس. أطلق الطبيب الروماني من أصل يوناني جالينوس ، الذي عاش في القرن الثاني ، على مرض hemicrania ، والذي يُترجم إلى "نصف الرأس". من هذا المصطلح جاءت كلمة "الصداع النصفي". في التسعينيات. في القرن العشرين ، وجد أن الصداع النصفي يرجع في الغالب إلى عوامل وراثية. تم اكتشاف عدد من الجينات المسؤولة عن انتقال الصداع النصفي عن طريق الوراثة.
تعتبر الأمراض الوراثية فريدة من نوعها من حيث أنها لا تعتمد على نمط حياة الشخص ، ولا يمكنك التأمين ضدها ببساطة عن طريق التوقف عن تناول الأطعمة الدهنية أو البدء في ممارسة التمارين في الصباح. تنشأ نتيجة طفرة ويمكن أن تنتقل من جيل إلى جيل.
مرض وراثي نادر يموت فيه الشخص بسبب عدم القدرة على النوم. حتى الآن ، لوحظ في 40 عائلة فقط حول العالم. يظهر الأرق القاتل عادةً بين 30 و 60 عامًا (غالبًا بعد 50 عامًا) ويستمر من 7 إلى 36 شهرًا. مع تقدم المرض ، يعاني المريض من اضطرابات نوم حادة بشكل متزايد ، ولا تساعده الحبوب المنومة. في المرحلة الأولى ، يصاحب الأرق نوبات هلع ورهاب ؛ وفي المرحلة الثانية ، تضاف الهلوسة وزيادة التعرق. في المرحلة الثالثة من المرض ، يفقد الشخص تمامًا قدرته على النوم ويبدأ في الظهور بمظهر أكبر من سنواته. ثم يتطور الخرف ويموت المريض عادة من الإرهاق أو الالتهاب الرئوي.

متلازمة الخدار - الجمدة ، التي تتميز بنوبات النوم المفاجئة واسترخاء عضلات الجسم ، لها أيضًا طبيعة وراثية وتنشأ من اضطرابات في مرحلة نوم حركة العين السريعة. إنه أكثر شيوعًا من الأرق العائلي المميت: في 40 من كل 100000 شخص ، بالتساوي بين الرجال والنساء. يمكن لأي شخص يعاني من النوم القهري أن ينام فجأة لبضع دقائق في منتصف النهار. تشبه "نوبات النوم" نوم حركة العين السريعة ويمكن أن تحدث كثيرًا ، حتى 100 مرة في اليوم ، مع أو بدون صداع يسبقها. غالبًا ما يتم استفزازها بسبب الخمول ، ولكن يمكن أن تحدث في أوقات غير مناسبة تمامًا: أثناء الاتصال الجنسي ، والرياضة ، والقيادة. الإنسان يستيقظ مرتاحا.
![]()
تتميز متلازمة يونر تان (UTS) في المقام الأول بحقيقة أن الأشخاص الذين يعانون منها يمشون في كل مكان. اكتشفه عالم الأحياء التركي يونر تان بعد دراسة خمسة أفراد من عائلة Ulas في ريف تركيا. في أغلب الأحيان ، يستخدم الأشخاص المصابون بـ SYT الكلام البدائي ولديهم فشل خلقي في الدماغ. في عام 2006 ، تم تصوير فيلم وثائقي عن عائلة Ulas بعنوان "Family Walking on All Fours". يصف تان الأمر بهذه الطريقة: "تشير الطبيعة الجينية للمتلازمة إلى خطوة عكسية في التطور البشري ، على الأرجح ناتجة عن طفرة جينية ، وهي العملية العكسية للانتقال من الرباعية (المشي على أربعة أطراف) إلى المشي على قدمين (المشي على طرفين) ). في هذه الحالة ، تتوافق المتلازمة مع نظرية التوازن المتقطع.

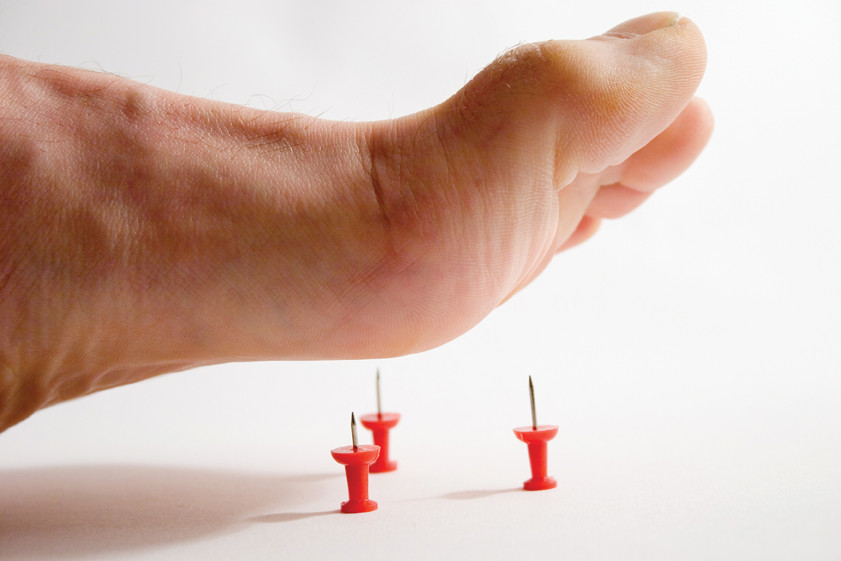
من أندر الأمراض في العالم: يتم تشخيص هذا النوع من الاعتلال العصبي لدى شخصين من بين مليون شخص. يحدث الشذوذ بسبب تلف الجهاز العصبي المحيطي الناتج عن زيادة الجين PMP22. العلامة الرئيسية لتطور الاعتلال العصبي الحسي الوراثي من النوع الأول هو فقدان الإحساس في اليدين والقدمين. يتوقف الشخص عن الشعور بالألم والشعور بتغير في درجة الحرارة ، مما قد يؤدي إلى نخر الأنسجة ، على سبيل المثال ، إذا لم يتم التعرف على كسر أو إصابة أخرى في الوقت المناسب. الألم هو أحد ردود أفعال الجسم التي تشير إلى أي "خلل" ، وبالتالي فإن فقدان حساسية الألم محفوف بالتأخر في الكشف عن الأمراض الخطيرة ، سواء كانت التهابات أو قرحًا.
يبدو الأشخاص الذين يعانون من هذا المرض غير العادي أكبر سناً بكثير من أعمارهم ، ولهذا يطلق عليه أحيانًا اسم "متلازمة بنجامين باتون العكسية". بسبب طفرة جينية وراثية ، وأحيانًا نتيجة استخدام بعض الأدوية في الجسم ، تتعطل آليات المناعة الذاتية ، مما يؤدي إلى فقدان سريع لمخزون الدهون تحت الجلد. الأنسجة الدهنية للوجه والرقبة الأطراف العلويةوالجذع ، مما يؤدي إلى ظهور التجاعيد والطيات. حتى الآن ، تم تأكيد 200 حالة فقط من حالات الحثل الشحمي التدريجي ، وهي تتطور بشكل رئيسي عند النساء. يستخدم الأطباء حقن الأنسولين وشد الوجه والكولاجين للعلاج ، لكنها مؤقتة فقط.

يُطلق على فرط الشعر أيضًا اسم "متلازمة بالذئب" أو "متلازمة أبرامز". إنه يصيب شخصًا واحدًا فقط من بين كل مليار شخص ، وقد تم توثيق 50 حالة فقط منذ العصور الوسطى. يتميز الأشخاص الذين يعانون من فرط الشعر بزيادة شعر الوجه والأذنين والكتفين. هذا بسبب انتهاك الروابط بين البشرة والأدمة أثناء تكوين بصيلات الشعر في جنين يبلغ من العمر ثلاثة أشهر. عادةً ، تخبر الإشارات من الأدمة الناشئة البصيلات شكلها. كما تشير البصيلات بدورها إلى طبقات الجلد أن هناك بصيلة واحدة بالفعل في هذه المنطقة ، وهذا يؤدي إلى حقيقة أن الشعر على الجسم ينمو على نفس المسافة تقريبًا من بعضها البعض. في حالة فرط الشعر ، تنكسر هذه الوصلات ، مما يؤدي إلى تكوين شعر كثيف جدًا في تلك الأجزاء من الجسم حيث لا ينبغي أن يكون.

إذا كنت قد سمعت من قبل عن إغماء الماعز ، فأنت تعرف تقريبًا ما يبدو عليه التوتر العضلي الخلقي - بسبب التشنجات العضلية ، يبدو أن الشخص يتجمد لفترة من الوقت. سبب التوتر العضلي الخلقي هو خلل وراثي: بسبب طفرة ، تتعطل قنوات الكلوريد في عضلات الهيكل العظمي. أنسجة العضلات "مشوشة" ، وهناك تقلصات اعتباطية واسترخاء ، ويمكن أن يؤثر علم الأمراض على عضلات الساقين والذراعين والفكين والحجاب الحاجز.

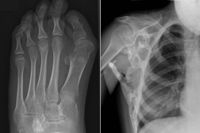
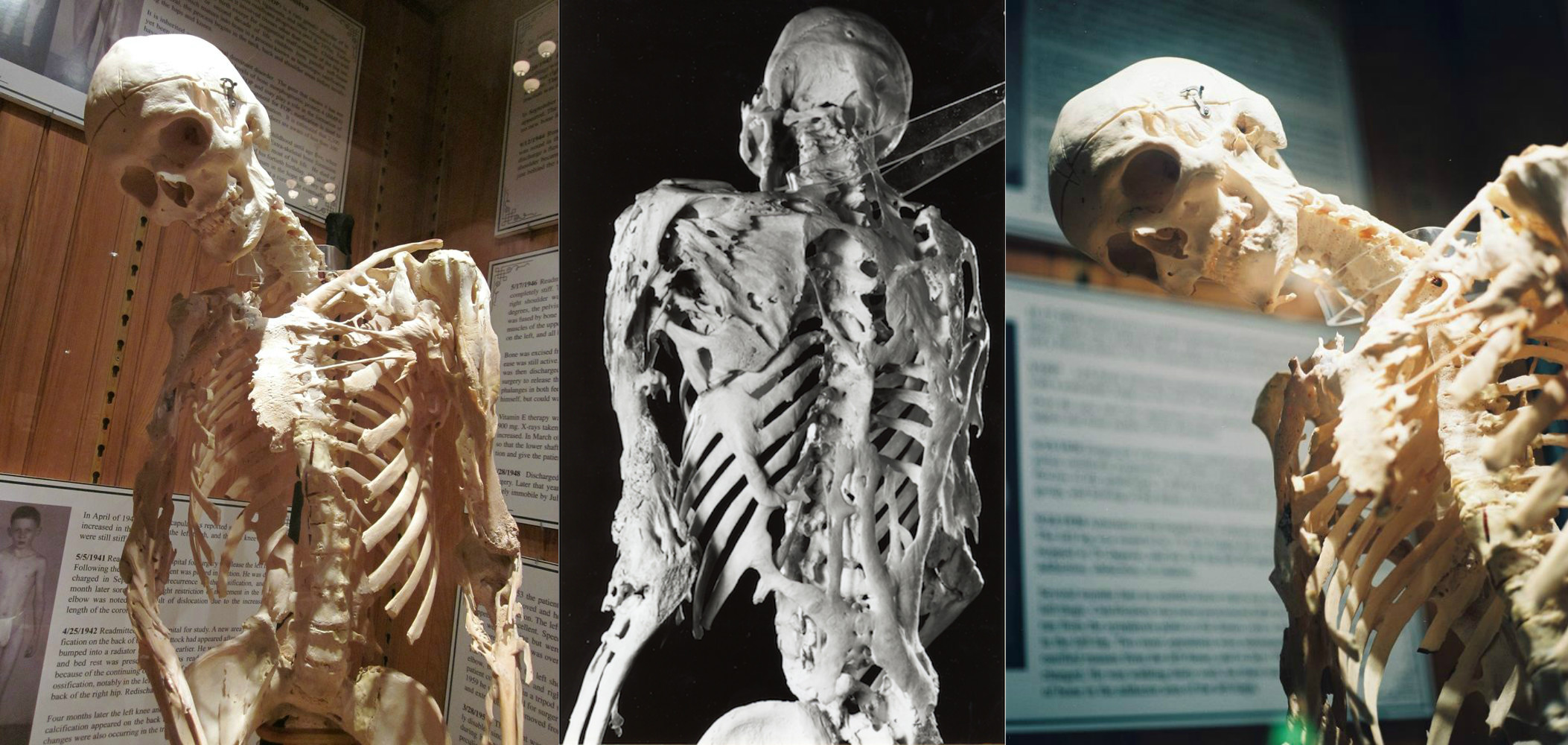
مرض وراثي نادر يبدأ فيه الجسم في تكوين عظام جديدة - عظام - في الأماكن الخطأ: داخل العضلات والأربطة والأوتار والأنسجة الضامة الأخرى. يمكن أن تؤدي أي إصابة إلى تكوينها: كدمات أو قطع أو كسر أو حقن عضلي أو عملية. لهذا السبب ، من المستحيل إزالة العظام: بعد الجراحة ، لا يمكن للعظام إلا أن تنمو أقوى. من الناحية الفسيولوجية ، لا تختلف العظام عن العظام العادية ويمكنها تحمل أحمال كبيرة ، لكنها ليست في المكان المناسب.
ينشأ FOP من طفرة في الجين ACVR1 / ALK2 الذي يشفر مستقبلات بروتين العظام. ينتقل إلى الإنسان بميراث أحد الوالدين إذا كان مريضًا أيضًا. يستحيل أن تكون حاملاً لهذا المرض: فالمريض إما مريض أو لا. حتى الآن ، يعتبر FOP من بين الأمراض المستعصية ، ولكن الآن هناك سلسلة ثانية من التجارب لعقار يسمى palovarotene ، والذي يسمح لك بمنع الجين المسؤول عن علم الأمراض ، قيد التنفيذ.
يتجلى هذا المرض الجلدي الوراثي في زيادة حساسية الشخص للأشعة فوق البنفسجية. ينشأ بسبب طفرة في البروتينات المسؤولة عن إصلاح تلف الحمض النووي الذي يحدث عند التعرض للأشعة فوق البنفسجية. تظهر الأعراض الأولى عادة في مرحلة الطفولة المبكرة (قبل 3 سنوات): عندما يكون الطفل في الشمس ، يصاب بحروق خطيرة بعد بضع دقائق فقط من التعرض للشمس. كما يتميز المرض بظهور النمش وجفاف الجلد وتغير لون الجلد. وفقًا للإحصاءات ، فإن الأشخاص المصابين بجفاف الجلد الصباغي أكثر عرضة للإصابة بالسرطان من غيرهم: في حالة عدم وجود تدابير وقائية مناسبة ، يصاب حوالي نصف الأطفال المصابين بجفاف الجلد الصباغي ببعض أنواع السرطان بحلول سن العاشرة. هناك ثمانية أنواع من هذا المرض متفاوتة الخطورة والأعراض. وفقًا للأطباء الأوروبيين والأمريكيين ، فإن المرض يحدث تقريبًا أربعة أشخاصمن بين مليون.

اسم فضولي لمرض ، أليس كذلك؟ ومع ذلك ، هناك أيضًا مصطلح علمي لهذا "التهاب اللسان التقشري". يظهر اللسان الجغرافي في حوالي 2.58٪ من الناس ، وغالبًا ما يكون للمرض خصائص مزمنة ويزداد سوءًا بعد تناول الطعام أو أثناء الإجهاد أو الإجهاد الهرموني. تتجلى الأعراض في ظهور بقع ملساء متغيرة اللون على اللسان ، تشبه الجزر ، ولهذا السبب تلقى المرض مثل هذا الاسم المستعار غير المعتاد ، وبمرور الوقت ، تغير بعض "الجزر" شكلها وموقعها ، اعتمادًا على أي من براعم التذوق الموجودة على اللسان يشفي ، وعلى العكس من ذلك ، يغضب.
اللسان الجغرافي غير ضار عمليًا ، إذا لم تأخذ في الاعتبار الحساسية المتزايدة للأطعمة الحارة أو بعض الانزعاج الذي يمكن أن يسببه. لا يعرف الطب أسباب هذا المرض ، ولكن هناك أدلة على الاستعداد الوراثي لتطوره.
ولادة طفل- أسعد حدث لكل زوجين. غالبًا ما تطغى الأفكار المقلقة على صحته ونموه السليم على انتظار لقاء مع الطفل. في معظم الحالات ، يتضح أن مخاوف الآباء الصغار تذهب سدى ، ولكن في بعض الأحيان يعامل القدر الطفل الذي لم يولد بعد بقسوة شديدة: لا يتلقى الطفل من الأم والأب لون الشعر وشكل العين وابتسامة حلوة فحسب ، بل أيضًا العديد من الأمراض الوراثية. .
وفقًا للإحصاءات الطبية ، فإن احتمال إنجاب طفل مصاب بمرض وراثي لكل أم حامل هو 3-5 ٪. على سبيل المثال ، احتمال إنجاب أطفال مصابين بمتلازمة داون هو 1: 700. أصعب التشخيص وقابل لمزيد من العلاج نادر ، ما يسمى بالأمراض اليتيمة: تكون العظم الناقص ، وانحلال البشرة الفقاعي ، ومتلازمة مينكس ، والشياخ ، وغيرها الكثير. كقاعدة عامة ، تشكل هذه الأمراض الوراثية الوراثية تهديدًا لحياة الطفل ، وتقلل بشكل كبير من مدتها وجودتها ، وتؤدي إلى الإعاقة. في بلدنا ، يعتبر مرض "نادر" مرضًا يحدث بمعدل تكرار 1: 10000.
أسباب الأمراض الوراثية
 كل خلية جسم الانسانيحمل رمزًا معينًا داخل الكروموسومات. في المجموع ، لدى الشخص 46 منهم: 22 منهم أزواج جسمية ، والزوج الثالث والعشرون من الكروموسومات مسؤول عن جنس الشخص. تتكون الكروموسومات بدورها من العديد من الجينات التي تحمل معلومات عنها خاصية معينةالكائن الحي. تحتوي الخلية الأولى التي تشكلت عند الحمل على 23 كروموسومًا للأم ونفس عدد الكروموسومات الأبوية. يؤدي خلل في الجين أو الكروموسوم إلى اضطراب وراثي.
كل خلية جسم الانسانيحمل رمزًا معينًا داخل الكروموسومات. في المجموع ، لدى الشخص 46 منهم: 22 منهم أزواج جسمية ، والزوج الثالث والعشرون من الكروموسومات مسؤول عن جنس الشخص. تتكون الكروموسومات بدورها من العديد من الجينات التي تحمل معلومات عنها خاصية معينةالكائن الحي. تحتوي الخلية الأولى التي تشكلت عند الحمل على 23 كروموسومًا للأم ونفس عدد الكروموسومات الأبوية. يؤدي خلل في الجين أو الكروموسوم إلى اضطراب وراثي.
هناك أنواع مختلفة من الاضطرابات الوراثية: عيب جين واحد ، وخلل في الكروموسومات ، وخلل معقد.
عيب جين واحديمكن أن ينتقل من أحد الوالدين أو كليهما. علاوة على ذلك ، كناقل الجينات المتنحية، قد لا يعرف أبي وأمي حتى عن مرضهم. تشمل هذه الأمراض الشيخوخة المبكرة ومتلازمة مينكس وانحلال البشرة الفقاعي وتكوين العظم الناقص. يسمى العيب الذي ينتقل مع الكروموسوم 23 مرتبط بالكروموسوم. يرث كل شخص كروموسوم X من والدته ، ولكن من والده يمكنه الحصول على كروموسوم Y (في هذه الحالة ، ولد ولد) أو كروموسوم X (تظهر فتاة). إذا تم العثور على جين معيب في كروموسوم X الخاص بالصبي ، فلا يمكن موازنته بواسطة كروموسوم X صحي ثانٍ ، وبالتالي هناك إمكانية لعلم الأمراض. يمكن أن ينتقل هذا العيب من الأم الحاملة للمرض أو يتشكل بشكل غير متوقع تمامًا.
عيب الكروموسوم- التغيير في هيكلها وعددها. في الأساس ، تتشكل هذه العيوب أثناء تكوين البويضات والحيوانات المنوية للوالدين ، ويحدث عيب في الكروموسومات في الجنين عندما تندمج هذه الخلايا. يتجلى مثل هذا المرض ، كقاعدة عامة ، في شكل اضطرابات خطيرة في النمو البدني والعقلي.
عيوب معقدةتنشأ نتيجة التعرض لجين أو مجموعة جينات من العوامل البيئية. لا تزال آلية انتقال هذه الأمراض غير مفهومة تمامًا. وفقًا لافتراضات الأطباء ، يرث الطفل من الوالد حساسية خاصة لـ عوامل معينة بيئة، تحت تأثير المرض يمكن أن يتطور في نهاية المطاف.
التشخيص في فترة ما قبل الولادة
 يمكن الكشف عن الأمراض الوراثية للأطفال حتى في فترة ما قبل الولادة. لذلك ، في الآونة الأخيرة ، في العديد من الاستشارات ، يتم إجراء اختبار يحدد مستوى هرمونات AFP وهرمون الاستروجين و hCG لجميع النساء بين الأسبوعين 18 من الحمل. يساعد في تحديد علم الأمراض النمائي للطفل بسبب عيوب الكروموسومات. وتجدر الإشارة إلى أن هذا الفحص يكشف فقط عن جزء من الاضطرابات الوراثية التصنيف الحديثالأمراض الوراثية نظام معقد يشمل حوالي ألفي مرض وحالة ومتلازمة.
يمكن الكشف عن الأمراض الوراثية للأطفال حتى في فترة ما قبل الولادة. لذلك ، في الآونة الأخيرة ، في العديد من الاستشارات ، يتم إجراء اختبار يحدد مستوى هرمونات AFP وهرمون الاستروجين و hCG لجميع النساء بين الأسبوعين 18 من الحمل. يساعد في تحديد علم الأمراض النمائي للطفل بسبب عيوب الكروموسومات. وتجدر الإشارة إلى أن هذا الفحص يكشف فقط عن جزء من الاضطرابات الوراثية التصنيف الحديثالأمراض الوراثية نظام معقد يشمل حوالي ألفي مرض وحالة ومتلازمة.
يجب على الآباء المستقبليين أن يضعوا في اعتبارهم أنه بناءً على نتائج هذا التحليل ، لا يتم تشخيص مرض معين ، ولكن يتم تحديد احتماله فقط ويتم اتخاذ قرار بشأن الحاجة إلى فحوصات إضافية.
فحص السائل الأمنيوسي- إجراء يقوم خلاله الطبيب ، باستخدام إبرة رفيعة وطويلة ، بسحب السائل الأمنيوسي ، ليخترق رحم المرأة من خلال جدار البطن. في السابق ، يتم إرسال المرأة لإجراء فحص بالموجات فوق الصوتية لتحديد وضع الجنين و افضل مكانإدخال الإبرة. في بعض الأحيان يتم إجراء الموجات فوق الصوتية أثناء إجراء بزل السلى.
تتيح لك هذه الدراسة التعرف على العديد من العيوب الصبغية ، وتحديد درجة تطور رئتي الطفل (إذا كان من الضروري الولادة قبل الموعد المحدد) ، وتحديد جنس الطفل بدقة (إذا كان هناك خطر من الأمراض المرتبطة جنس معين). تستغرق دراسة السائل الناتج عدة أسابيع. عيب هذا الإجراء هو أنه يمكن إجراؤه في عمر الحمل الذي يزيد عن 16 أسبوعًا ، مما يعني أنه لم يتبق للمرأة سوى القليل من الوقت لاتخاذ قرار بشأن الإجهاض. بالإضافة إلى ذلك ، على عكس الثلث الأول من الحمل ، فإن الإجهاض في مثل هذا الوقت الطويل هو إجراء خطير للغاية لكل من الصحة الجسدية والعقلية للمرأة. تتراوح مخاطر الإجهاض التلقائي بعد هذه الدراسة من 0.5 إلى 1٪.
بمساعدة دراسة المشيمة (النسيج المحيط بالجنين في بداية الحمل) ، من الممكن أيضًا تحديد الاضطرابات الوراثية في الجنين ، بما في ذلك تشخيص الأمراض النادرة إلى حد ما ، مثل انحلال البشرة الفقاعي وتكوين العظم الناقص. خلال هذا الإجراء ، يُدخل الطبيب أنبوبًا رفيعًا عبر المهبل في رحم المرأة. يتم شفط قطع من الزغابات المشيمية من خلال أنبوب ثم إرسالها للتحليل. هذا الإجراء غير مؤلم ويمكن إجراؤه في وقت مبكر من الأسبوع التاسع من الحمل ، وستكون نتائج الدراسة جاهزة في غضون يوم إلى يومين. على الرغم من المزايا الواضحة ، فإن هذا الإجراء ليس مطلوبًا بشدة بسبب ارتفاع مخاطر الإجهاض التلقائي (2-3 ٪) واضطرابات الحمل المختلفة.
مؤشرات دراسة المشيماء وبزل السلى هي:
- عمر الأم الحامل أكثر من 35 عامًا ؛
- عيوب صبغية في أحد الوالدين أو كليهما ؛
- ولادة طفل مصاب بعيوب صبغية في زوجين ؛
- الأمهات الحوامل اللواتي في عائلاتهن مصابات بأمراض مرتبطة بالكروموسوم X.
إذا أكدت الدراسات وجود اضطراب وراثي ، فسيتعين على الوالدين ، بعد موازنة جميع الإيجابيات والسلبيات ، أن يتخذوا ربما الخيار الأصعب في حياتهم: الحفاظ على الحمل أو إنهائه ، منذ علاج الأمراض الوراثية عند هذا الحد. المرحلة للأسف مستحيلة.
التشخيص بعد الولادة
 يمكن تشخيص الأمراض الوراثية الوراثية النادرة على أساس الاختبارات المعملية. منذ عدة سنوات ، في جميع مستشفيات الولادة ، في اليوم الخامس بعد ولادة الطفل ، تم إجراء فحص حديثي الولادة ، يتم خلاله تشخيص عدد من الأمراض الوراثية النادرة: بيلة الفينيل كيتون ، قصور الغدة الدرقية ، التليف الكيسي ، الجالاكتوز في الدم ، والغدة الكظرية. متلازمة.
يمكن تشخيص الأمراض الوراثية الوراثية النادرة على أساس الاختبارات المعملية. منذ عدة سنوات ، في جميع مستشفيات الولادة ، في اليوم الخامس بعد ولادة الطفل ، تم إجراء فحص حديثي الولادة ، يتم خلاله تشخيص عدد من الأمراض الوراثية النادرة: بيلة الفينيل كيتون ، قصور الغدة الدرقية ، التليف الكيسي ، الجالاكتوز في الدم ، والغدة الكظرية. متلازمة.
يتم تشخيص الأمراض الأخرى على أساس الأعراض والعلامات التي يمكن أن تحدث في فترة حديثي الولادة وسنوات عديدة بعد الولادة. تظهر أعراض انحلال البشرة الفقاعي وتكوين العظم الناقص في معظم الحالات بعد الولادة مباشرة ، وغالبًا ما يتم تشخيص الشيخوخة المبكرة فقط في عمر 2-3 سنوات.
من الصعب جدًا على طبيب الأطفال العادي التعرف على الأمراض النادرة ، فقد لا يلاحظ الطبيب ببساطة أعراضها خلال موعد منتظم. لهذا السبب يجب أن تكون الأم منتبهة جدًا لطفلها وتهتم بالعلامات المهددة: المهارات الحركية التي تجاوزت العمر ، وظهور النوبات ، وزيادة الوزن بشكل غير كافٍ ، واللون غير الطبيعي ورائحة حركات الأمعاء. أيضا ، يجب أن يكون سبب الإنذار هو الزيادة الحادة أو التباطؤ في عملية نمو الطفل ، وهذا قد يشير إلى وجود مرض مثل التقزم. عندما تظهر مثل هذه الأعراض ، يجب على الآباء بالتأكيد استشارة الطبيب ، والإصرار على إجراء فحص شامل للطفل ، لأن التشخيص في الوقت المناسب للأمراض الوراثية واختيار برنامج العلاج الصحيح يمكن أن يساعد في الحفاظ على صحة الطفل وأحيانًا على حياته.
كيف يتم علاج الأمراض الوراثية؟
على الرغم من أن معظم الأمراض الوراثية لا يمكن علاجها ، فإن الطب الحديث قادر على زيادة متوسط العمر المتوقع للأطفال المرضى بشكل كبير ، فضلاً عن تحسين جودته. حتى الآن ، هذه الأمراض ليست جملة ، ولكنها طريقة حياة تسمح للطفل بالتطور بشكل طبيعي ، بشرط أن يتم تلقي العلاج اللازم: تناول الأدوية ، والجمباز ، والوجبات الغذائية الخاصة. علاوة على ذلك ، كلما كان من الممكن التشخيص في وقت مبكر ، يتم تنفيذ علاج الأمراض الوراثية بنجاح.
في الآونة الأخيرة ، تم استخدام طرق العلاج قبل الولادة بشكل متزايد: بمساعدة الأدوية وحتى العمليات الجراحية.
مرض الطفل هو اختبار صعب لجميع أفراد الأسرة. في ظل هذه الظروف ، من المهم جدًا أن يقوم الآباء بدعم الأقارب والتواصل مع الأمهات والآباء الآخرين الذين يجدون أنفسهم في وضع مماثل. يتم مساعدة هذه العائلات بشكل كبير من قبل مجتمعات مختلفة من الآباء والأمهات الذين لديهم أطفال يعانون من أمراض وراثية نادرة.
كيف نمنع الأمراض الوراثية؟
سيساعد التخطيط السليم للحمل ، والذي ينصب تركيزه الرئيسي على الوقاية من الأمراض الوراثية ، على تجنب ولادة طفل مريض. يجب على الآباء المعرضين للخطر بالتأكيد زيارة أحد علماء الوراثة:
- سن الوالدين -35 سنة وما فوق ؛
- إنجاب طفل أو أكثر مرض وراثي;
- أمراض نادرة في الأزواج أو أقاربهم ؛
- قلق الأزواج بشأن إنجاب طفل سليم.
بناءً على بيانات الفحص الطبي ، بالإضافة إلى معلومات حول التاريخ العائلي ، والأمراض التي يعاني منها الأقارب ، ووجود عمليات إجهاض وإجهاض ، يقوم استشاري الجينات بحساب احتمال إنجاب طفل بمرض وراثي. يحدث أن يتخلى الزوجان اللذان لديهما فرصة كبيرة في ولادة طفل مريض عن هذه الخطط في هذا الاتحاد ، ومع شركاء آخرين يكتسبون أطفالًا أصحاء تمامًا.
فتيات! لنقم بإعادة النشر.
بفضل هذا ، يأتي إلينا الخبراء ويقدمون إجابات لأسئلتنا!
أيضا ، يمكنك طرح سؤالك أدناه. الناس مثلك أو الخبراء سيقدمون إجابة.
شكرًا ؛-)
كل الأطفال الأصحاء!
ملاحظة. هذا ينطبق على الأولاد أيضا! يوجد المزيد من الفتيات هنا ؛-)
هل أعجبتك المادة؟ الدعم - إعادة النشر! نحن نحاول من أجلك ؛-)
هذا منشور! الأصل مأخوذ من eka_tyryshkina في عيوب الطبيعة: الأشخاص المصابون بأمراض نادرة.
لقد مرضت هنا في اليوم الآخر ، كما هو الحال دائمًا ، لأننا نحتاج فقط للذهاب للزيارة ، أنا مريض! توليلي يتفاعل مرضي مع الحدث المخطط له ، أو لشيء آخر ، لكن من الجيد أنه لا يتفاعل مع العمل. بشكل عام ، مرضي ليس بسيطا)
والآن مريض في المنزل ساعة متأخرة، بعد أن أعدت بالفعل جميع الحالات ، وأعدت قراءتها وتمريرها عبر جميع المواقع المثيرة للاهتمام ، وفجأة ، وبشكل غير متوقع بالنسبة لي ، قررت التعرف على أندر الأمراض على هذا الكوكب ، وكما تعلم ، هناك الكثير من الأشياء المثيرة والصادمة. !!!
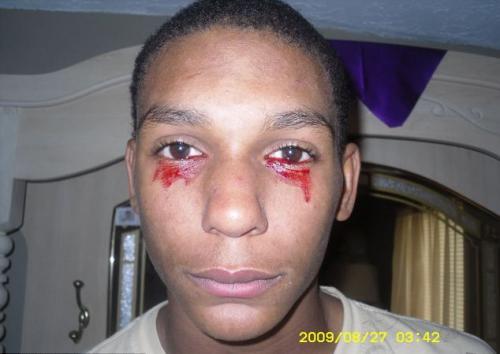
الهيمولاكريا
("دموع الدم") تحدث في شخص واحد من بين كل مليون شخص ، ويبدأ الدم ، بدلاً من السائل المسيل للدموع ، في التدفق من العين فجأة ، ويمكن أن يستمر هذا لمدة ساعة تقريبًا. خلال النهار ، يذرف المريض دموعًا دموية من 3 إلى 20 مرة.
السبب الدقيق لهذا المرض غير مفهوم تمامًا ، وبالتالي لا يمكن علاجه. لا يزال الأخصائيون الطبيون يطرحون نسخًا مفادها أن hemolacria هو أحد أمراض الدم أو الأورام.
في الصورة - 15 عاما كالفينو إنمان(تينيسي ، الولايات المتحدة الأمريكية)

متلازمة مصاص الدماء
مع التشخيص "متلازمة مصاص الدماء"
(خلل التنسج الأديم الظاهر) في العالم لا يوجد سوى 7 آلاف شخص.
بالإضافة إلى الجلد الشاحب المميت والأنياب الحادة (في حالة عدم وجود جزء من الأسنان) ، يكون لدى المرضى شعر متناثر ورقيق ، وتقل القدرة على التعرق ، وبالتالي فإن أجسامهم عرضة لارتفاع درجة الحرارة. تظهر الأعراض في مرحلة الطفولة ولكن يمكن اكتشاف المرض بالفعل في مرحلة الحمل باستخدام الاختبارات الجينية.
يُجبر الأولاد على ارتداء نظارات داكنة وواقي من الشمس عندما يخرجون لأنهم لا يمكن أن يكونوا تحت أشعة الشمس المباشرة.في الوقت نفسه ، يظل النمو البدني والنشاط الحركي طبيعيين.المرض نفسه غير قابل للشفاء ، ويمكن تصحيح الأعراض فقط. على وجه الخصوص ، من الممكن استعادة الشكل الطبيعي للأسنان.
تم تشخيص مرض سيمون في الطفولة. عندما كانت ماندي حامل للمرة الثانية ، تم تحذيرها من أن الطفل الثاني يمكن أن يصاب بنفس المرض.ومع ذلك ، نما سيمون وتطور بشكل جيد ، لذلك خاطر والديه بهذه المخاطرة.
يقول الأولاد "بعض الأطفال يسخرون من مظهرنا لكن أصدقائنا يعتقدون أنه رائع"
في الصورة - سيمون (13 عامًا) وجورج (11 عامًا) كولين (سوفولك ، المملكة المتحدة).

فرط الشعر
("متلازمة الذئب") هو مرض يتجلى في نمو الشعر المفرط الذي لا يتميز بهذه المنطقة من الجلد ، ولا يتوافق مع الجنس والعمر. تم تسجيل أكثر من أربعين مريضًا بقليل في جميع أنحاء العالم ، لذلك الطريقة الأكثر ملاءمة بالنسبة لهم لكسب المال هي إظهار قبحهم ... يتقدمون بطلب إلى كتاب غينيس للأرقام القياسية ليصبحوا مشهورين ويكسبون المال ... نجح الصيني Yu Zhenhuang بنسبة مائة بالمائة - بفضل شعره الفائق ، أسس فرقة الروك الأكثر شهرة في بلاده وأصبح مليونيرا.
من غير المعروف سبب حدوث هذه الطفرة. ولم يطور أحد حتى الآن علاجًا لفرط الشعر. يمكن لأخصائيي التجميل إزالة الشعر فقط لفترة طويلة بما فيه الكفاية ...
في الصورة - 6 سنوات نات ساسوفان(تايلاند) ، 2007

في الصورة - يو جينهوانغ البالغ من العمر 33 عامًا (الصين) ، اشعر رجل في العالم

داء الفيل
("متلازمة المتقلبة" ، داء الفيل ، داء الفيل ، داء الفيل) - زيادة في حجم أي جزء من الجسم بسبب النمو المؤلم للجلد والأنسجة تحت الجلد. هناك ما يقرب من 120 شخصًا في العالم يعانون من هذا المرض المستعصي ...
وأشهر مريض هو "رجل الفيل" - جوزيف ميريك. في عام 1980 ، قدم المخرج ديفيد لينش فيلمًا عن البريطاني الشهير ، والذي تم ترشيحه لجائزة الأوسكار في ثمانية ترشيحات ... كان الفيلم عن كرامة الإنسان ... تم إنشاء مكياج جون هيرت ، الذي لعب دور ميريك. على أساس تلك المقدمة في المستشفى الملكي في لندن ، جثة جوزيف ميريك الكحولية. استغرق تراكبه اليومي الممثل 12 ساعة في اليوم ...
في الصورة - يبلغ من العمر 35 عامًا ماندي سيلارز(بريطانيا العظمى)

شذوذ جيني يتكون من متسارع شيخوخة الجسم ,
- بروجيريا- تنقسم إلى أطفال (متلازمة هاتشنسون) وكبار (متلازمة ويرنر). لأول مرة ، تمت مناقشة متلازمة الشيخوخة المبكرة منذ 100 عام. وليس من المستغرب أن تحدث مثل هذه الحالات مرة واحدة من بين 4-8 ملايين طفل. Progeria (من المؤيد اليوناني - سابقًا ، gerontos - الرجل العجوز) هو مرض وراثي نادر للغاية يؤدي إلى تسريع عملية الشيخوخة بحوالي 8-10 مرات. ببساطة ، طفل يتراوح عمره بين 10 و 15 عامًا في عام واحد. طفل يبلغ من العمر ثماني سنوات يبدو عمره 80 عامًا - ببشرة جافة مجعدة ورأس أصلع ... يموت هؤلاء الأطفال عادة في سن 13-14 بعد عدة نوبات قلبية وسكتات دماغية على خلفية تصلب الشرايين التدريجي وإعتام عدسة العين والزرق ، خسارة كاملة للأسنان ، إلخ. وقليل منهم فقط يعيشون حتى 20 عامًا أو أكثر.
الآن فقط 42 حالة لأشخاص مصابين بالشيخوخة المبكرة معروفة في العالم ... من بين هؤلاء ، يعيش 14 شخصًا في الولايات المتحدة ، و 5 في روسيا ، والباقي في أوروبا ...
حاليا ، هناك العديد من المنظمات التي تقدم المساعدة للمسنين الصغار وأسرهم. هناك مواقع على الإنترنت مخصصة لهذه المشكلة بالذات ، بعضها فتحه أطباء أو الأخصائيين الاجتماعيين، آخرون - أسر المرضى.
في الصورة - ليون بوت البالغ من العمر 24 عامًا



38 سنة رجل الشجرة
أصبح ديدي كوسوارا ، الذي يعيش في جزيرة جاوة بإندونيسيا ، مشهورًا في جميع أنحاء العالم بسبب فيروس الورم الحليمي البشري ، والذي يؤدي عادةً إلى ظهور الثآليل الصغيرة ، ولكن في حالة الإندونيسي ، فقد تشوه أطرافه بشكل يتعذر التعرف عليه.
كانت مشكلة ديدي أنه كان يعاني من اضطراب وراثي نادر يمنع جهاز المناعة لديه من منع نمو الثآليل. لذلك ، استطاع الفيروس "السيطرة على الآلية الخلوية لخلايا جلده" ، وأعطاها أوامر بإنتاج كمية كبيرة من المادة القرنية التي تكونت منها. كان لدى ديدي أيضًا نسبة منخفضة من الكريات البيض في دمه.


مرض الفراشة
انحلال البشرة الفقاعي في شكل مفرط التنسج هو مرض وراثي يظهر في الأيام الأولى من الحياة. في الواقع ، جلد المولود رقيق للغاية لدرجة أن أي لمسة تؤدي إلى تقرحات وبثور. المناطق البارزة تعاني أكثر من غيرها: المرفقين والركبتين والقدمين واليدين. القرحة الناتجة ، التي يخرج منها الجلد في طبقات ، لا تلتئم لفترة طويلة ، ويتم إطلاق السوائل منها. بعد تشكيل ندبة قرمزية كبيرة.
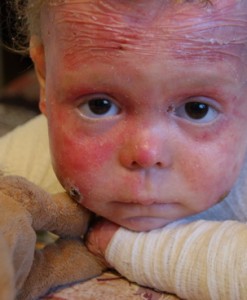
لا يوجد علاج لهذا المرض ، يمكن فقط تخفيف الأعراض. منذ وقت ليس ببعيد ، انتشرت قصة ليزا كونيجل ، التي كانت تعيش مع انحلال البشرة الفقاعي منذ ما يقرب من عشر سنوات ، في جميع أنحاء روسيا. عدة مرات في اليوم ، تحتاج إلى ضمادات وعلاج بالمراهم والمواد الهلامية المضادة للميكروبات. بالإضافة إلى ذلك ، طوال 9 سنوات ، كانت ليزا مصحوبة بالألم.
متلازمة حورية البحر
واحدة من أندر الحالات الشاذة في التطور هي صفارات الإنذار ، والتي يطلق عليها شعبيا "متلازمة حورية البحر". مع هذا العيب ، يولد الأطفال حديثو الولادة بأرجل مقسمة ، على غرار ذيل السمكة. لديهم كلية واحدة فقط وليس لديهم أعضاء تناسلية. بسبب أضرار جسيمة اعضاء داخليةوعادة ما يموت هؤلاء الأطفال بعد فترة وجيزة. يحدث المرض في واحد من كل 100.000 مولود جديد. على مدار سنوات المراقبة ، تمكن ثلاثة أطفال فقط من البقاء على قيد الحياة. كان أحدهم شيلو بيبين.
ولدت شيلوه عام 1999 وأصبحت أشهر طفل مصاب بمتلازمة حورية البحر. على مدى السنوات العشر التي تمكنت فيها من العيش ، كان لديها الآلاف من الأصدقاء حول العالم الذين دعموا الفتاة ووالدتها. حاولت شيلوه أن تعيش حياة كاملة - فقد ذهبت ، مثل كل الأطفال العاديين ، إلى المدرسة ، وحضرت دروس الرقص ، وذهبت إلى المتنزهات الترفيهية. أصبحت الفتاة مشهورة بعد مشاركتها في عرض أوبرا وينفري. قامت شركة Learning Chanel بعمل عدة أفلام عنها ، ومئات المواقع على الإنترنت مخصصة لها.

قصة شيلوه هي قصة مذهلة عن معجزة. طفل قاتل طوال طفولته من أجل البقاء. طفلة صغيرة تعرف كيف تستمتع كل يوم بالرغم من مرض عضال.
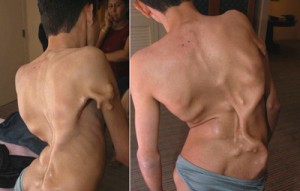
مرض مونهايمر
يعتبر خلل التنسج الليفي مرض نادر للغاية. الإحصاءات الرسمية هي كما يلي: مريض واحد لكل 2،000،000 شخص. يحدث مرض مونهايمر نتيجة طفرة جينية ويتجلى عند الولادة في عيوب خارجية. ثني أصابع القدم الكبيرة والعمود الفقري. علم الأمراض يؤدي إلى الإعاقة والموت المبكر. في الأماكن التي يجب أن تحدث فيها العمليات المضادة للالتهابات ، يبدأ نمو العظام في التكون ، وهذا هو سبب تسمية المرض "بمرض الهيكل العظمي الثاني".
يمكن أن تؤدي أي كدمة ، حتى ولو كانت بسيطة ، إلى ظهور زجاج في المنطقة المصابة ، ولا يوجد حتى الآن علاج رسمي لمرض مميت. طور العلماء دواء يمكنه نظريا محاربة المرض. ومع ذلك ، لم يتم إجراء الدراسات السريرية اللازمة بعد. للأسف ، من الصعب جدًا إجراؤها - لا يوجد أكثر من 600 شخص مصاب بمرض مونهايمر في جميع أنحاء العالم.
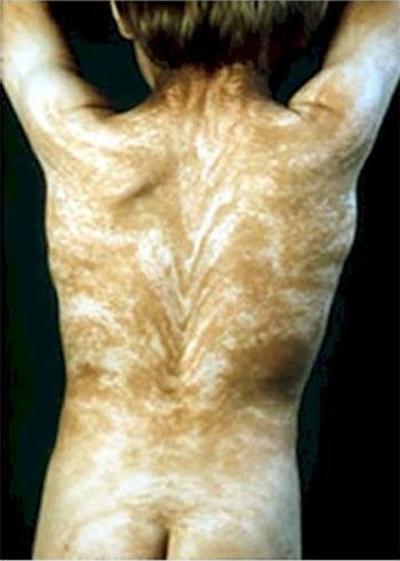
ظاهرة "Line Blaschko"تتميز بوجود خطوط غريبة في جميع أنحاء الجسم. خطوط Blaschko هي نمط غير مرئي مضمن في الحمض النووي. ويصبح مظهر المرض هو وضوح هذا النمط.
عادةً ما يكون النمط الموجود على الظهر على شكل حرف V ، وعلى الصدر والبطن والجانبين - على شكل حرف S.
قد يكون سبب المرض هو الفسيفساء. على أي حال ، فإن ظهور خطوط Blaschko لا علاقة له بالجهاز العصبي والعضلي واللمفاوي البشري.
مرض آخر غير طبيعي acantokeratoderma، أو "متلازمة الجلد الأزرق". يمكن أن يكون لدى الأشخاص الذين يعانون من هذا التشخيص بشرة زرقاء أو نيلية أو برقوقية أو شبه أرجوانية.في الستينيات من القرن الماضي ، عاشت عائلة كاملة من الأشخاص "الأزرقين" في كنتاكي. كانوا معروفين باسم Blue Fugates. تم نقل هذه الميزة من جيل إلى جيل.


يعاني حوالي 6٪ من سكان العالم من أمراض نادرة ، ويستمر هذا العدد في الازدياد. جميع الأمراض الفريدة من نوعها لها طبيعة مختلفة ، ولكن الغالبية العظمى من الأمراض الهائلة مرتبطة بالتشوهات الجينية والالتهابات.